Метод - симметрия - потенциальная функция
Cтраница 1
Метод симметрии потенциальных функций позволяет не только перечислить оптимальные способы формирования молекулярных кристаллов, но и количественно охарактеризовать относительную вероятность структурных классов. С этой целью, учитывая число степеней свободы, которое имеют молекулы и их агломераты при том или ином способе наложения, вводят эмпирические коэффициенты, пропорциональные вероятностям соответствующих стадий построения кристалла. [1]
Метод симметрии потенциальных функций позволяет ограничить количество возможных политипов, не вводя никаких предположений о конкретном строении слоя, а следовательно, и без требования плотнейшего наложения слоев. [2]
Итак, метод симметрии потенциальных функций дает адекватную интерпретацию общей картины строения молекулярных ( в частности, органических) кристаллов. Однако применительно к каждому конкретному веществу его предсказательные возможности явно недостаточны. Этот метод не чувствует индивидуальности молекул, поскольку концентрирует внимание лишь на их симметрии и симметрии поля межмолекулярных сил. Один из эффективных способов учета специфики молекул, их геометрических особенностей, определяющих конкретную кристаллическую структуру - это использовать аппарат ван-дер-ваальсовых радиусов, позволяющий представить органический кристалл как плотную упаковку объемных тел. [3]
Более общий и более обоснованный ( математически и физически) подход к проблеме открывает метод симметрии потенциальных функций [41-43], сущность которого заключается в следующем. [4]
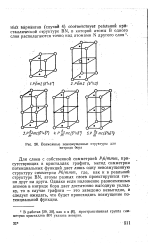 |
Возможные невоэмущенные структуры для нитрида бора. [5] |
Для слоев с собственной симметрией Pfi / mmm, присутствующих в кристаллах графита, метод симметрии потенциальных функций дает лишь одну невозмущенную структуру симметрии Рб / mmm, где, как и в реальной структуре BN, атомы разных слоев проектируются точно друг на друга. Однако если наложение разноименных атомов в нитриде бора дает достаточно выгодную укладку, то в случае графита - это заведомо невыгодно, и следует ожидать, что будет происходить возмущение потенциальной функции. [6]
В настоящей работе описано теоретическое определение оптимальной структуры хлорида меди, проведенное последовательным применением метода симметрии потенциальных функций, принципа плотной упаковки и энергетического расчета. [7]
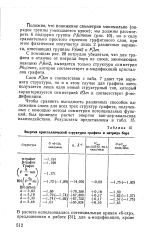 |
Энергия кристаллической структуры графита и нитрида бора. [8] |
Чтобы сравнить выгодность различных способов наложения слоев для всех трех структур графита, полученных с помощью метода симметрии потенциальных функций, был проведен расчет энергии ван-дер-ваальсова взаимодействия. [9]
Итак, на основании проведенных расчетов по крайней мере в 12 случаях из рассмотренных 21, реальные кристаллические структуры удается с очевидностью отнести к одному из подклассов, выводимых с помощью метода симметрии потенциальных функций. В четырех случаях это отнесение потребовало дополнительного указания на слабую выраженность слоев, что отмечалось в записи подклассов с помощью скобок. Наконец, в пяти случаях обнаруженные подклассы потребовали дополнительного предположения о перестройке слоев в процессе образования кристалла. [10]
Хотя объектом нашего исследования были слоистые ван-дер-ваальсовые структуры, полученные данные могут быть использованы и для дальнейшего изучения молекулярных кристаллов. Действительно, схема применения метода симметрии потенциальных функций к молекулярным кристаллам включает анализ наложения слоев; эту последнюю стадию для всех случаев симметрии молекул в основном отражают таблицы, обсуждаемые ниже. [11]
Наряду с этим, поскольку существует связь между подклассом и физическими свойствами, было бы важно предсказать, какие именно подклассы могут встретиться среди молекулярных кристаллов. Такую возможность дает разработанный одним из нас [4, 5] метод симметрии потенциальных функций, который позволяет в рамках определенных постулатов теоретически вывести допустимые для молекулярных кристаллов структурные классы и подклассы и оценить степень вероятности их реализации. [12]
По-видимому, возникновение таких структур обусловлено тем, что первоначально возникшие группировки молекул перестраиваются в процессе кристаллизации. Подобный путь образования кристалла не предусматривается исходными постулатами метода симметрии потенциальных функций и поэтому естественно, что подкласс / г-дихлорбензола не вытекает из проведенного теоретического вывода. [13]
Однако даже для такого сравнительно простого случая, каким являются слоистые ван-дер-ваальсовы кристаллы, перебор всех возможных вариантов относитель ного расположения структурных фрагментов с подсчетом для каждого из этих вариантов коэффициента плотности упаковки или энергии ван-дер-ваальсова взаимодействия представляется чрезвычайно громоздким. Существенное упрощение задачи дают полученные нами результаты применения метода симметрии потенциальных функций, которые позволяют значительно сузить диапазон допустимых расположений слоев. [14]
В настоящей работе описано теоретическое определение оптимальной структуры хлорида меди, проведенное последовательным применением метода симметрии потенциальных функций, принципа плотной упаковки и энергетического расчета. Для дисульфида молибдена мы ограничились применением метода симметрии потенциальных функций; этого было достаточно для предсказания возможных слоистых политипов. [15]
Страницы: 1 2