Модель - потенциал
Cтраница 2
В качестве модели потенциала возьмем МТ-потенциал, построенный в промежутке между сферами и сферически-симметричный внутри сфер. В этом разделе удобнее считать, что сферы не перекрываются. Имея такую картину металла, можно вполне строго считать электроны между сферами свободными, а влияние сфер учесть, рассматривая рассеяние свободных, электронов на них. При этом весь эффект от сфер содержится; в граничных условиях, наложенных на волновые функции на границе сфер. Эта процедура очень похожа на то, что мы делали в методе псевдопотенциала, однако при рассмотрении; d - зон сложность их электронной структуры находит свое отражение в сложном характере рассеяния. [16]
В предыдущем разделе был приведен обзор ряда теоретических результатов по межмолекулярным силам. Используя эти результаты, попытаемся построить модели потенциалов, которые достаточно правильно отражают поведение реальных межмолекулярных сил и вместе с тем довольно просты для их математического описания. До самого последнего времени эти условия были почти несовместимы, однако применение быстродействующих ЭЦВМ сделало возможными расчеты прямым численным интегрированием по крайней мере второго вириального коэффициента. Эти расчеты настолько просты, что не нуждаются в математическом упрощении. Применение ЭЦВМ до некоторой степени решило одну проблему, но, к сожалению, при этом возникла другая, более важная проблема. Она состоит в том, что почти бесполезно рассматривать потенциалы с двумя или тремя ( независимо выбираемыми) параметрами. В этом случае экспериментальные данные не позволяют получить надежную информацию о параметрах для слишком гибких потенциалов. [17]
Условия для е и с получены в соответствии с теорией дисперсионных лондоновских сил, и в действительности они не являются независимыми. Тем не менее приведенные правила следует рассматривать как полуэмпирические, так как модель потенциала получается путем простого сложения короткодействующих и дальнодеиствующих членов, что не является строгим в теоретическом смысле. [18]
Результаты для двух пар потенциалов совершенно различны. Может быть сделан основной вывод о значительном уменьшении неаддитивных поправок, а также о том, что неаддитивные поправки чувствительны к выбору модели потенциала. Очевидно, что успех наших знаний о тройном взаимодействии зависит от развития наших представлений о парном взаимодействии. Неаддитивные поправки также ставят под сомнение использование данных о кристалле для определения потенциалов двойного взаимодействия. [19]
Потенциал ( п - 6) стал особенно популярен после того, как Леннард-Джонс проинтегрировал выражение для второго вири-ального коэффициента с помощью быстросходящегося степенного ряда, позволяющего получить достаточно хорошие результаты даже при использовании небольших настольных счетных мащин. Это обстоятельство явилось немаловажным в период, предшествовавший бурному развитию ЭВМ. Модель потенциала оказалась успешной и широко используется до сих пор. [20]
Рассмотрено влияние строения двойного слоя на закономерности фотоэмиссии электронов с поверхности металлического электрода в раствор. Выяснена зависимость фототока от ф - потенциала и других параметров, характеризующих двойной слой, на основании чего предложен метод определения указанных параметров по отклонению вольтамперной характеристики электрода ( или зависимости фототока от частоты света) от известного закона пяти вторых. С помощью модели потенциалов нулевого радиуса рассмотрено влияние дискретности адсорбционного слоя при малых числах заполнения и найдена зависимость фототока от поверхностной концентрации адсорбата. [21]
Выбор формы потенциала Ф для взаимодействия сложных молекул с адсорбентом на основании таких расчетов, по-видимому, пока практически невозможен. Вместе с тем определение зависимости потенциальной энергии Ф межмолекулярного взаимодействия сложной молекулы с адсорбентом от ее ориентации у поверхности на основании экспериментальных адсорбционных данных также практически невозможно из-за недостаточной чувствительности термодинамических характеристик адсорбции к модели этой зависимости. Кроме того, такие эмпирические определения формы потенциала Ф необходимо было бы проводить для каждой интересующей нас системы в отдельности. Вместе с тем модели потенциалов, учитывающие зависимость межмолекулярного взаимодействия от ориентации молекул, применяемые при расчетах свойств разреженных газов, по-видимому, не годятся для расчетов свойств адсорбционных систем, так как адсорбционные свойства более чувствительны к геометрическому строению молекулы, чем свойства объемных газов. [22]
Они нашли, что поправка составляет 8 % для потенциала ( 9 - 6) и 5 % для потенциала ( 12 - 6) в достаточно широком интервале температур. Грабен, Презент и Маккаллох [118] тоже рассчитали обменный вклад в АС. Но они использовали более сложный потенциал, основанный на одноэлектронной модели Гаусса [87], которая включает вклады первого и второго порядков в Аи3, обм, хотя соответствующий им парный потенциал ( 12 - 6) содержит только вклады первого порядка в и0бм - Во всех случаях можно сделать следующий общий вывод, что АС0бм и АСДИСперс имеют противоположные знаки и сравнимы по величине, поэтому суммарная неаддитивная поправка АС мала. К сожалению, квантовые результаты оказываются очень чувствительными к выбору модели потенциала двойного взаимодействия. Этот вопрос обсуждается в разд. [23]
Необходимо проверить предположение о том, что значительная часть воды находится в связанном состоянии в той области макроиона, где величина электростатического потенциала высока. Если это так, то кинетической единицей в растворах многих электролитов является ион со сравнительно более прочно связанными молекулами воды. Определение параметра а, характеризующего в видоизмененном уравнении Дебая - Хюккеля размер иона, подтверждает это предположение. В любом случае концепция ионного связывания, которую можно объяснить, исходя из описанной ранее модели гибридного потенциала, требует присутствия воды между ионизован ным макроионом, несущим электрический заряд, и связан - ными противоионами. Только при этом условии удается четко разграничить влияние электростатического взаимодействия и влияние ассоциации ионов. [24]
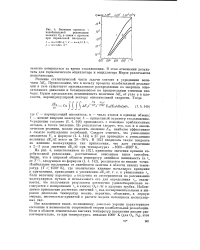 |
Значение времени т колебательной релаксации молекул 02 в смеси с аргоном при нормальной плотности. [25] |
На рис. 4, заимствованном из 182 ], приведены значения времени колебательной релаксации, рассчитанные описанным выше способом. Видно, что в широкой области температур линейная зависимость In t, от T - i, следующая из формулы ( I. Если величину параметра а получить из экспериментов по рассеиванию молекулярных пучков и использовать его для определения tK, оказывается, что полученные результаты достаточно удовлетворительно согласуются с измерениями - к в смесях 02 Аг в ударных трубах. Наблюдаемое превышение расчетных значений тк над экспериментальными в широком диапазоне температур можно, вероятно, объяснить, в первую очередь, неточностью принятой в расчете модели потенциала молекулярных сил. [26]
Опубликованные коэффициенты активности электролитов при средней ыоляльной концентрации ( определенной вышеописанным методом) учитывают как взаимодействие между ионами, так и взаимодействие ионов с растворителем. Необходимо проверить предположение о том, что значительная часть воды находится в связанном состоянии в той области макроиона, где величина электростатического потенциала высока. Если это так, то кинетической единицей в растворах многих электролитов является ион со сравнительно более прочно связанными молекулами воды. Определение параметра я, характеризующего в видоизмененном уравнении Дебая - Хюккеля размер иона, подтверждает это предположение. В любом случае концепция ионного связывания, которую можно объяснить, исходя из описанной ранее модели гибридного потенциала, требует присутствия воды между ионизованным макроионом, несущим электрический заряд, и связанными противоионами. Только при этом условии удается четко разграничить влияние электростатического взаимодействия и влияние ассоциации ионов. [27]
Страницы: 1 2