Образование - изобутан
Cтраница 2
Из представлений о простой полимеризации следовало бы ожидать, что димеризация этилена будет давать бутен-1 или - 2, которые могут гидрироваться до w - бутана. Следовательно, образование изобутана можно объяснить при допущении изомеризации бутена-1 или - 2 в изобутилен с последующей гидрогенизацией последнего в изобутан. [16]
Одним из важнейших типов молекулярных перегруппировок, протекающих с участием ионов карбония, является изомеризация парафинов. Наибольший интерес представляет образование изобутана из н-бутана, происходящее в присутствии хлористого или бромистого алюминия. При комнатной температуре равновесие устанавливается, когда смесь состоит приблизительно из 80 % изобутана и 20 % н-бутана. [17]
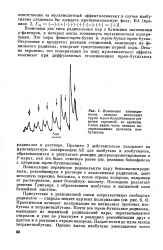 |
Изменение интенсивности сигнала метильных групп mpem - бутилбензоата ( отмечен стрелкой во времени. Слева видно часть сигнала поляризованных протонов изо-бутилена. [18] |
Покинувшие первичную радикальную пару бензоилоксиради-калы, рекомбинируя в растворе с аналогичным радикалом, дают исходную перекись бензоила, а после отрыва водорода, например от растворителя, дают бензойную кислоту. Последняя разлагает реактив Гриньяра с образованием изобутана и магниевой соли бензойной кислоты. [19]
Хотя раздельно окись алюминия и окись цинка не являются сколько-нибудь удовлетворительными катализаторами изосин-теза, каждый из них может использоваться как эффективный промотор для окиси тория. Добавка окиси алюминия увеличивает газообразование ( особенно образование изобутана), а окись цинка увеличивает выход жидких углеводородов. Поэтому логично было предположить, что катализаторы, состоящие из окиси алюминия и окиси цинка, могут служить заменителями окиси тория. В графах 6 - 9 рис. 11 показаны результаты опытов, проводившихся с такими катализаторами, приготовленными методом обращенного осаждения, объемная производительность которых оказалась, однако, значительно ниже, чем окисноториевых. Выход продуктов изосннтеза при 150 am можно увеличить снижением объемной скорости газа па 50 %, однако это приводит к дополнительному уменьБгепию объемной производительности. Сравнение результатов, приведенных в графах 8 и 9 ( рис. 11), показывает пебходп-мость относительно высокого содержания окиси алюминия для уменьшения выхода спиртов. [20]
В частности, выход бутан-бутиленовой фракции возрастает с 3 4 до 4 4 вес. Это увеличение протекает в основном за счет большего образования изобутана ( с 1 8 до 2 5 вес. Последнее обстоятельство указывает на предпочтительное протекание вторичных реакций при крекинге очищенного сырья. [21]
Изоалкаиы при одинаковых условиях гидрогенизации ( температура, давление) расщепляются быстрее, чем алканы. Например, изооктан расщепляется примерно в 20 раз быстрее, чем н-гептан, с образованием почти чистого изобутана. [22]
Нормальные бутилены - смесь бутена-1 ( 8 %) и бутена-2 ( 92 %), не изменяясь заметно при 400 С, подвергаются довольно глубоким превращениям при 500 С. Основные реакции: распад, изомеризация и образование насыщенных углеводородов и, что особенно интересно, образование изобутана. [23]
В условиях каталитического крекинга протекают многие реакции изомеризации. Эти реакции совершенно отличны от неравновесных скелетных перегруппировок, которые имеют место только при крекинге индивидуальных молекул и ведут, например, к образованию изобутана как основного продукта при крекинге более высокомолекулярных парафиновых углеводородов нормального строения. Истинные реакции изомеризации, стремящиеся к состоянию равновесия, протекают при крекинге олефиновых и ароматических углеводородов и обычно являются вторичными реакциями, следующими за первичными реакциями крекинга, в результате которых образуются непредельные углеводороды. Несмотря на то, что в присутствии крекирующих катализаторов иногда наблюдается изомеризация насыщенных углеводородов ( нафтеновых или парафиновых), значение этой реакции в обычных условиях крекинга невелико. Хиндин, Облед и Миллс [53] установили, что при температуре 150 и продолжительности опыта 1 час в присутствии алюмосиликатного слабо гидратированного катализатора ( добавлено 0 5 % воды) наблюдается изомеризация парафиновых углеводородов С6, или С7, содерясащих третичный углеродный атом. [24]
Были выделены также сравнительно небольшие количества продуктов, образовавшихся по реакции переноса водорода: 7 - 9 % метилциклопентана, 6 - 7 % циклогексана, 8 - 12 % триметилпен-танов. Метилциклопентан получается в результате изомеризации образующегося в качестве промежуточного продукта реакции циклогексиль-ного катиона до того, Как он превращается в циклопарафин по схеме, аналогичной схеме образования изобутана из бутенов-1 и - 2 по реакции переноса водорода. [25]
Этот механизм объясняет более быстрое избирательное насыщение третичных непредельных углеводородов. Так как третичные ионы более стабильны, образование их происходит значительно быстрее, что обеспечивает более высокую концентрацию их на поверхности и, следовательно, более высокую скорость образования изобутана, изопентана и других подобных продуктов, что и наблюдается в действительности. [26]
Предложенная схема носит несколько спорный характер. Данные радиометрического анализа продуктов гидрокрекинга дурола, содержащего 14СН3, показали почти статистическое распределение радиоактивности по всем углеродным атомам ( в том числе, что особенно необычно, и в ароматических углеводородах С8 - С9), что делает представление об образовании изобутана посредством сбора метильных групп в длинную боковую цепь маловероятным. Это подтверждается и крайне малым количеством бензола в продуктах гидрокрекинга. Очевидно этот процесс протекает сложнее, чем его можно представить приведенной выше схемой. [27]
Предложенная схема носит несколько спорный характер. Данные радиометрического анализа продуктов гидрокрекинга дурола, содержащего UGH3, показали почти статистическое распределение радиоактивности по всем углеродным атомам ( в том числе, что особенно необычно, и в ароматических углеводородах С8 - С9), что делает представление об образовании изобутана посредством сбора метильных групп в длинную боковую цепь маловероятным. Это подтверждается и крайне малым количеством бензола в продуктах гидрокрекинга. Очевидно этот процесс протекает сложнее, чем его можно предста7 вить приведений выше схемой. [28]
Реакции алкильных групп алкилпроизводных соединений ароматического ряда полностью определяются размером этих групп. Ароматические углеводороды, содержащие метальные или этильные заместители, гидрируются либо частично, либо полностью с образованием нафтенов, а дезалкилирование почти не происходит. Тетра-метилбензол и аналогичные алкилпроизводные данного гомологического ряда подвергаются деалкилированию с отщеплением метильных групп от ароматических колец и образованием изобутана. Расщепления же бензольных колец практически не происходит. Эта необычная реакция описана Эганом, Ланглуа и Уайтом [11], которые обнаружили, что боковые цепи, содержащие пропильные, бутильные и пентильные группы, подвергаются простому дезалкилированию и превращаются в парафины. Дезалкилирование затрагивает и более длинные боковые цепи, но при этом одновременно происходит циклизация, в результате которой продукты гидрокрекинга обогащаются тетралинами и инданами. [29]
Реакция последовательного дегидрирования протекает на катализаторе, а стадия циклизации является термической. Наряду с основной реакцией циклизации протекают побочные реакции. Наиболее важными из них являются крекинг, приводящий к образованию легких углеводородов от Cj до С5, полимеризация части непредельных с образованием небольших количеств высококипящих углеводородов и кокса; кроме того, проходят реакции гидрирования олефинов ( выделяющимся при дегидроциклизации водородом), скелетной изомеризации исходного гексана и продуктов крекинга, приводящей к образованию изобутана, изопентана, изогексанов, изобути-ленов и изоамиленов. Если в н-гексане присутствуют примеси изогексанов или метилциклопентана, реакция скелетной изомеризации приобретает важное значение и предшествует циклизации. [30]
Страницы: 1 2 3