Описание - электронная структура - молекула
Cтраница 1
Описание электронной структуры молекулы С2Н2 в рамках метода локализованных МО исходит из линейности молекулы. [1]
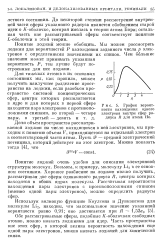 |
График вероятности нахождения одного электрона внутри сфер радиуса R для атома Не. [2] |
Понятие лоджий очень удобно для описания электронной структуры молекул. [3]
Применяя представление о резонансе структур к описанию электронной структуры молекул, следует всегда помнить, что отдельные резонансные структуры не выражают физически реальных состояний молекулы. [4]
Метод ВС, как и метод МО, представляет собой вариант орбитального описания электронной структуры молекулы. В нем, как и в методе МО, может быть формулировано я-электронное приближение. Последовательное и достаточно полное развитие методов ВС и МО позволяет установить, что в отношении вида получаемых волновых функций оба метода по существу имеют общую основу. [5]
В некоторых случаях, без учета резонанса структур, в рамках метода ВС может получаться качественно неправильное описание электронной структуры молекулы. Так, для бензола ни одна из двух классических формул Кекуле не отражает реальной симметрии молекулы, а также ее физических и химических свойств. Длина связи углерод - кислород в нем равна 0 115 - нм, тогда как длина нормальной двойной связи СО ( в кетонах) равна 0 122 нм, а расчетная длина тройной связи СО - 0 110 нм. [6]
В некоторых случаях, без учета резонанса структур, в рамках метода ВС может получаться качественно неправильное описание электронной структуры молекулы. Так, для бензола ни одна из двух классических формул Кекуле не отражает реальной симметрии молекулы, а также ее физических и химических свойств. Другой пример-диоксид углерода СОг - Длина связи углерод-кислород в нем равна 0 115 нм, тогда как длина нормальной двойной связи СО ( в кетонах) равна 0 122 нм, а расчетная длина тройной связи СО-0110 нм. [7]
Заканчивая изложение идеи о резонансе структур, важно отметить, что специфическим для этого подхода к описанию электронной структуры молекулы является не образование линейной комбинации волновых функций ( это весьма общий способ построения, типичный для квантовой механики вообще), а выбор функций для образования линейной комбинации. Существенным здесь является также и то, что выбираемые для образования линейных комбинаций волновые функции являются приближенными многоэлектронными волновыми функциями молекулы в целом. [8]
Эти экспериментальные данные позволяют провести описание электронной структуры молекулы при помощи двухцентровых орбиталей метода ЛМО. [9]
Развитие исследований процессов атомных и молекулярных столкновений, рассеяния, сопровождаемого реакциями, а также предиссо-циационных явлений вызвало в последнее время оживление интереса к теории валентных связей как к методу получения кривых и поверхностей потенциальной энергии ( см., например, [21, 210]), В традиционной формулировке теории валентных связей молекулярная волновая функция записывается как произведение волновых функций составляющих атомов. Обычно это позволяет получать качественно правильную картину диссоциационных процессов. Точность вычислений может быть улучшена учетом в волновой функции дополнительных структур. Однако, прибегая к многоструктурному методу валентных связей точно так же, как и в многоконфигурационных методах Хартри - Фока, мы вынуждены расстаться с простой картиной, достигаемой в рамках модели независимых электронов. В данном разделе излагается модельный подход для описания электронной структуры молекул, который может рассматриваться как простое обобщение и метода молекулярных орбиталей, и метода валентных связей. Речь идет о применении этих двух методов в сочетании друг с другом, что, с одной стороны, устраняет многие их недостатки, а с другой - сохраняет простую физическую картину электронной структуры, достигаемую при использовании модели независимых электронов. [10]
Помимо многообразия физических экспериментов по ЯМР широкому использованию этого метода способствует также большое разнообразие магнитных ядер, важных с химической точки зрения. Эти ядра важны и при исследованиях в неорганической химии. Кроме того, значительный интерес представляет спектроскопия ЯМР на ядрах многих металлов. Измерение этих постоянных позволяет проверить надежность квантовомеханических методов, используемых для описания электронной структуры молекул. Таким образом, на сегодняшний день спектроскопия ЯМР является необходимым методом для всех областей науки, будь то биология, химия или физика. [11]
Страницы: 1