Определение - концентрация - элемент
Cтраница 2
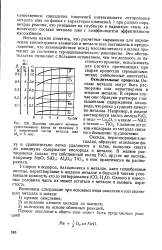 |
Влияние среднего времени существования ванны на величину J и химический состав металла шва ( d 5 мм. [16] |
Весьма важно отметить, что расчетные выражения для определения концентрации элементов в металле шва составлены в предположении, что взаимодействие между массами металла и шлака протекает до состояния термодинамического равновесия. Это обстоятельство позволяет с большим основанием, чем это делалось до настоящего времени, использовать для расчета протекающих при сварке процессов термодинамические равновесные константы. Оксиды металла могут быть растворимы или нерастворимы в жидком металле. [17]
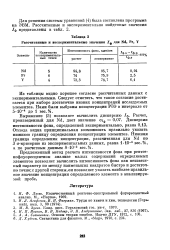 |
Рассчитанные и экспериментальные значения Гф для Nd, Pr, Y. [18] |
Отсюда видна принципиальная возможность правильно указать нижнюю границу определения концентрации элементов. Нижняя граница определения концентрации, рассчитанная для Nd по 3 a - критерию из экспериментальных данных, равна 1 - 10 - 4 вес. [19]
Описанное выше явление ослабления эмиссии кальция примесями применялось для определения концентрации тушащих элементов. [20]
Всякая задача спектрального анализа в конечном итоге сводится к определению концентрации элемента в пробе по результатам измерения интенсивностей его спектральных линий в плазме источника. Связь между этими двумя величинами может быть достаточно точно предсказана лишь в простейших случаях. Допустим, что анализируемое вещество равномерно поступает в плазму разряда и в результате каких-то процессов, например диффузии или конвекции, уходит из плазмы в окружающую атмосферу. [21]
Пламенные фотометры и спектрофотометры ( табл. 8) служат для определения концентраций элементов в растворах. Эмиссионный метод заключается в фотометри-ровании пламени, в которое вводится в распыленном виде анализируемый раствор. Атомно-абсорбционный метод основан на изменении интенсивности резонансной линии спектра за счет поглощения ее энергии атомами определяемого элемента, образующимися в пламени горелки при введении в него распыленного анализируемого раствора. [22]
Задачу можно решить, если использовать метод, основанный на определении концентрации элемента в образце путем графической или численной интерполяции между результатами измерений двух ближайших эталонов, один из которых содержит меньше определяемого элемента, а другой больше. [23]
Как известно, для большинства применяемых инструментальных методов анализа величина погрешности определения концентрации элемента в пробе зависит от близости состава анализируемой пробы и эталонных ( точнее образцовых) растворов, по которым построен гра-дуировочный график. Чтобы погрешность определения содержания элементов в пробе была по возможности меньше, образцовые растворы должны быть близки по своему составу к анализируемым пробам. Это требование в рассматриваемом случае осуществить практически невозможно, так как природные воды отличает сложность и непостоянство состава. Поэтому применение низкоселективных методов для анализа этих объектов ведет к высоким значениям погрешности анализа а часто и к несопоставимости результатов. [24]
Таким образом, интенсивность эмиссионной спектральной линии может быть использована в качестве аналитического сигнала для определения концентрации элемента. Коэффициент а в уравнении (11.17) является сугубо эмпирической величиной, зависящей от условий процесса. Поэтому в АЭС решающее значение имеет правильный выбор условий атомизации и измерения аналитического сигнала, включая градуировку по образцам сравнения. [25]
Физический смысл, вкладываемый в выражение концентрационная чувствительность линий - вполне ясен: величина К характеризует ошибку в определении концентрации элемента, которую вызывает та или иная вариация, или ошибка из ерения интенсивности линии. Что же касается количественного определения К. Различие между этим выражением и приведенным в тексте не имеет принципиального значения; однако, для практических целен в отношении фотографических и визуальных методов рациональнее применять выражение, приведенное в тексте, а для фотоэлектрических методов характеризовать АГ как АГД / / ДС. [26]
Опытный спектроскопист, систематически выполняющий анализы, при строгом соблюдении аналитических условий часто может обеспечить поразительную точность при определении концентраций элементов обоими методами. Вследствие наложения спектральных линий этими методами можно обнаружить несуществующие элементы, и, чтобы избежать таких ошибок, необходима высокая тщательность. Для каждого элемента результаты анализа необходимо проконтролировать по нескольким линиям. [27]
При этом с учетом многих факторов проанализировано влияние полной погрешности аналитического сигнала и погрешности воспроизведения цветной реакции на погрешность определения концентрации элемента и на полную погрешность фотометрического анализа. Здесь же показано, что в массовом фотометрическом анализе применение общего для партии проб заранее приготовленного реагента, содержащего все необходимые для осуществления цветной реакции компоненты, улучшает воспроизводимость результатов фотометрического анализа и сокращает его трудоемкость. [28]
Что касается второй из указанных возможных причин снижения сопротивления отрыву по границам зерен, - изменения состава твердого раствора в приграничных зонах зерен, - то отсутствие достаточно локальных прямых методом определения концентрации элементов в тонких приграничных зонах в течение длительного времени не позволяло достаточно определенно установить наличие таких изменений. [29]
При Т 2 - l ( f К, когда время реакции 4) перестает лимитировать горение, требуется учет каждой реакции в отдельности из (14.11), а также из (14.15) - (14.23) для определения концентраций элементов и скорости выделения энергии. Так как формулы типа (13.27) и (13.37) здесь весьма громоздки из-за наличия большого числа резонансов, приведем табл. 13 из [462], где приведены значения NA аи как функции температуры. [30]
Страницы: 1 2 3