Особенность - поведение - молекула
Cтраница 1
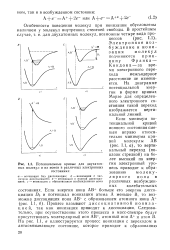 |
Потенциальные кривые для двухатомных молекул и их ионов в различных электронных состояниях. [1] |
Особенности поведения молекул при ионизации обусловлены наличием у молекул внутренних степеней свободы. Электронное возбуждение и ионизация молекул подчиняются принципу Франка - Кондона - за время электронного перехода межъядерное расстояние не изменяется. На диаграмме потенциальной энергии в форме кривых Морзе для определенного электронного состояния такой переход изображается вертикальной линией. [2]
С эффектами Яна-Теллера связывают и ряд особенностей поведения молекул в биологических системах. [3]
При очень малых и очень больших давлениях газа на коэффициентах переноса сказываются особенности поведения молекул в разреженном и концентрированном газе по сравнению с идеальным. При очень низких давлениях газа ( р - С 1 am) коэффициенты переноса определяются в соответствии с лучевым характером перемещения молекул. Большая концентрация молекул при больших давлениях газа приводит к заметному силовому взаимодействию молекул, которое оказывает влияние на коэффициенты переноса. [4]
При очень больших давлениях ( р 1 атм) на переносе в газах сказывается силовое взаимодействие молекул. Особенности поведения молекул могут сказываться на переносе и при обычных давлениях, но при состояниях газов, близких к насыщенному пару, когда возможна ассоциация молекул. С понижением температуры и повышением давления по мере приближения состояния газа к насыщенному пару поведение газа все в большей мере отклоняется от свойств идеального газа. [5]
Уже это позволило понять многие особенности поведения молекул, в первую очередь циклических и оптически деятельных. В основу конформационных представлений положен установленный экспериментально факт, что пространственные взаимоотношения между непосредственно не связанными друг с другом нейтральными атомами определяются не столько их объемами, зависящими от атомных радиусов, сколько эффективными, или ван-дер-ваальсовыми, объемами. Эти объемы, получившие в последние годы название конформационных, гораздо больше атомных ( например, атомный радиус водорода равен 0 030 нм, а конформационный - 0 120 нм), и именно ими определяется относительное расположение в пространстве отдельных частей молекул, если только на их взаимоотношениях не сказываются какие-либо другие еще более сильные взаимодействия. В частности, пространственное расположение атомов в молекулах алканов и циклоалканов определяется преимущественно конформационными объемами близлежащих, ш друг с другом не связанных атомов водорода. При сближении этих атомов на расстояния, несколько превышающие сумму их ван-дер-ваальсовых радиусов, между ними возникают силы отталкивания. Когда расстояния между несвязанными атомами равны или близки к этой сумме, силы отталкивания резко возрастают. Дальнейшее сближение или перекрывание ван-дер-ваальсовых радиусов может привести к неустойчивости молекулы и даже к ее разрушению. Под влиянием сил отталкивания все атомы водорода в молекуле стремятся расположиться как можно дальше друг от друга. [6]
Но эти характеристики в некоторой части справедливы и для разветвленных полимеров и для полимерных сеток. Их возникновение следует отнести не к особенностям поведения молекул в целом, а к линейным отрезкам цепей, заключенным между разветвлениями, или к самим разветвлениям, а также к отрезкам цепей, заключенным между узлами пространственной сетки. [7]
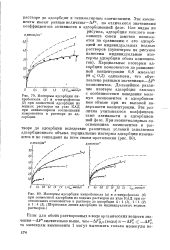
Парциальные изотермы адсорбции компонентов до равновесной концентрации 0 8 ммолъ / кг ( в 0 3) одинаковы, что обусловлено равными значениями - AF компонентов. Дальнейшее различие изотерм адсорбции связано с особенностями поведения молекул компонентов в адсорбционном объеме при их высокой поверхности концентрации. Эти различия учитываются коэффициентами активности в адсорбционной фазе. [9]
Свойства а - и я-орбит позволяют приближенно рассматривать поведение я-электронов независимо от поведения а-электронов; я-элек-троны как бы образуют внешнюю оболочку молекулы и двигаются в. Свойства о-связей в молекулах с сопряженными и несопряженными связями одинаковы и особенности поведения молекул с сопряженными связями определяются особенностями делокализованных я-орбит. [10]
Свойства 0 - и я-орбит позволяют приближенно рассматривать поведение я-электронов независимо от поведения а-электронов; я-электро-ны как бы образуют внешнюю оболочку молекулы и двигаются в поле атомных остовов, экранированных более прочно связанными 0 - электронами. Свойства а-связей в молекулах с сопряженными и несопряженными связями одинаковы и особенности поведения молекул с сопряженными связями определяются особенностями делокализованных я-орбит. [11]
Свойства а - и я-орбит позволяют приближенно рассматривать поведение я-электронов независимо от поведения о-электронов; я-элек-троны как бы образуют внешнюю оболочку молекулы и двигаются в: поле атомных остовов, экранированных более прочно связанными а-электроиами. Свойства о-связей в молекулах с сопряженными и несопряженными связями одинаковы и особенности поведения молекул с сопряженными связями определяются особенностями делокализоеанных я-орбит. [12]
Глубокое понимание конформационного поведения молекулы гидразина, а, следовательно, в значительной степени и насыщенных его производных, возможно лишь на основе квантово-механических расчетов. Современное состояние как вычислительной техники, так и методов расчета, не позволило пока получить данные для сложных производных; именно это и заставляет нас подробно рассматривать саму шестиатомнув молекулу гидразина, для которой многочисленные расчеты были проведены. Ценность таких расчетов, впрочем, далеко не одинакова. Трудные для понимания на основе классических стереохи-мических представлений особенности поведения молекулы гидразина, содержащей вицинальнне неподеленнне пары, оказываются столь же трудными для воспроизведения методами полуэмпирических расчетов. Сравнение результатов расчета потенциальных кривых внутреннего вращения методами CNDO, INDQ и NDDO показало, что при полуэмпирических расчетах барьеров внутреннего вращения в молекулах с соседними гетероатомами, обладающими неподеленными парами, необходимо применение таких методов, которые подобно NDDO гарантируют адекватное неэмпириче ским методам описание двухцентрового электронного взаимодействия, в особенности между орбиталями неподеленных пар. Принимая во внимание, что большинство полуэмпирических методов вообще непригодно для целей конформационного анализа [20], к результатам, полученным этими методами, следует относиться с осторожностью. [13]
Страницы: 1