Молекулярно-орбитальный подход
Cтраница 1
Молекулярно-орбитальный подход с учетом конфигурационного взаимодействия приводит к лучшим результатам, чем метод валентных связей. При межъядерных расстояниях, близких к равновесному значению, из волновой функции конфигурационного взаимодействия вычитается небольшая часть ионного вклада простой молекулярно-орбитальной волновой функции, а в пределе изолированных атомов - весь этот вклад. [1]
Долгое время считалось, что молекулярно-орбитальный подход имеет существ, недостаток: мол. Электронная плотность, отвечающая каждой орбитали, более или менее равномерно распределена по всему объему молекулы. [2]
Таким образом, мы видим, что молекулярно-орбитальный подход является безусловно шагом вперед по сравнению с обычной теорией кристаллического поля. Почему же в таком случае конкретные расчеты по методу кристаллического поля проводятся значительно чаще, чем по методу молекулярных орбит. Дело в том, что надежные численные методы расчета порядка следования уровней и расстояний между ними для второго метода практически пока не разработаны. Главным образом эти трудности связаны с введением дополнительных неизвестных параметров в методе молекулярных орбит - параметров смешивания орбиталей аир, отчего понижается надежность расчетов и при сравнении теории с экспериментом приходится пользоваться только качественными соображениями даже в таком вопросе, как порядок следования энергетических уровней, не говоря уже о величинах расщеплений и обобщении теории на случай низкосимметричного окружения переходных ионов лигандами. [3]
Теория ароматичности дает еще одно доказательство плодотворности молекулярно-орбитального подхода к оценкам строения органических соединений. [4]
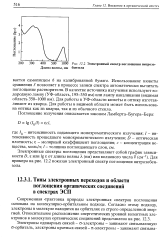 |
Электронный спектр поглощения нитрозо-бензола. [5] |
Современная - трактовка природы электронных спектров поглощения основана на молекулярно-орбитальном подходе. Согласно этому подходу, электроны в молекуле находятся на орбиталях со строго определенной энергией. [6]
Этот результат имеет общий характер: если исходить из заданного базисного набора, то молекулярно-орбитальный подход с полным учетом конфигурационного взаимодействия приводит к таким же результатам, как и метод валентных связей с полным учетом конфигурационного взаимодействия. [7]
При возбуждении электрона в свободной молекуле HF с орбитали 2ра на орбиталь 3sa получается состояние 12, которое обладает резко выраженными ионными свойствами. Таким образом, как и в методе валентных связей, при молекулярно-орбитальном подходе получается, что стабильность водородной связи обусловливается влиянием ионного состояния. В то же время связь Зхст-орбитали с ионным состоянием не является достаточно простой, а поэтому лучше рассматривать стабильность водородной связи с точки зрения влияния одной ( или нескольких) низколежащей возбужденной орбитали точно так же, как это сделано в книге [22] ( стр. Данный подход подчеркивает отличие водородной связи от дисперсионного взаимодействия, которое обусловливается коллективным воздействием всех возбужденных состояний соответствующей симметрии. [8]
Другое важное отличие теории валентных связей от теории молекулярных орбиталей состоит в том, что в этих теориях по-разному учитываются веса ионных структур. Ионные состояния появляются при учете резонанса различных валентных состояний. При молекулярно-орбитальном подходе ионные состояния принимаются во внимание с самого начала, причем так, что они даже переоцениваются. [9]
Члены в скобках зависят от координат только одного электрона, а члены, по которым проводится двойное суммирование, описывают двухэлектронные отталкивательные взаимодействия. Для небольших систем ( с числом электронов порядка десяти) удается провести прямые численные расчеты. Большинство расчетов систем сколько-нибудь значительного размера начинается с использования волновой функции в приближении независимых частиц. При этом чаще всего используется молекулярно-орбитальный подход теории самосогласованного поля Хартри - Фока. Расчеты по методу валентных связей применительно к большим молекулам используются гораздо реже. [10]
Страницы: 1