Положение - другой атом
Cтраница 1
Положения других атомов были получены из разностного синтеза Фурье, который был построен по измеренным и рассчитанным структурным факторам, найденным из уточненного положения атома таллия. [1]
Необходимо точное измерение интенсивностей и точное определение положений других атомов, так как атомы водорода могут выявиться только в случае устранения всех мешающих эффектов. [2]
 |
Расположение ато мов кремния ( большие кружки и углерода ( маленькие кружки в плоскости ( 1120 некоторых политипных структур карбида кремния. [3] |
Указанные / обозначения политипных структур простых веществ применимы и для описания политипных структур химических соединений. В этом случае элементарные слои структуры А, В и С будут уже не моноатомными, а полиатомными, и буквами Л, В и С в данном случае характеризуют позиции атомов одного сорта в элементарном слое, а положения других атомов в этом слое по отношению к указанным выше являются фиксированными. [4]
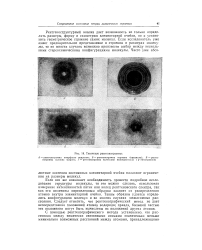 |
Типичные рентгенограммы. [5] |
Если все же возникает необходимость провести подробное исследование структуры молекулы, то это можно сделать, использовав измерение интенсивностей пятен или колец рентгеновского спектра, так как эти величины определенным образом зависят от распределения атомов внутри элементарной ячейки. Таким образом удается определить конфигурацию молекул и во многих случаях межатомные расстояния. Следует отметить, что рентгенографический метод не дает непосредственно положений атомов водорода; правда, большей частью-эти положения могут быть вычислены из положений других атомов. [6]
Если все же возникает необходимость провести подробное исследование структуры молекулы, то это можно сделать, использовав измерение интенсивностей рентгеновских интерференции, так как эти величины определенным образом зависят от распределения атомов внутри элементарной ячейки. Таким образом удается определить конфигурацию молекулы и во многих случаях межатомные расстояния. Следует отметить, что рентгеновский метод не дает возможности непосредственно определять положение атомов водорода; правда, эти положения могут быть вычислены из положений других атомов. [7]
Если рассмотреть в объеме элементарного кристалла ближайшее окружение какого-либо атома ( или атома данного типа в кристаллическом соединении), окажется, что число таких атомов и расстояния до них от рассматриваемого будут одни и те же, какой бы атом не был принят за центральный. При мысленном переносе атома ( или элементарной ячейки, состоящей из нескольких атомов) на любое расстояние, кратное межатомному расстоянию а, новое положение атома или ячейки точно совпадет с положением другого атома ( ячейки) в кристаллической решетке. Это свойство называют дальним порядком расположения атомов. [8]
В некоторых случаях таким путем удается сразу выявить кристаллическую структуру. Если молекула содержит только один тяжелый атом, то почти все фазы будут определяться именно этим атомом. Если, кроме того, этот тяжелый атом находится в частном положении, так что все три его координаты определяются симметрией, то можно прямо найти вероятные знаки всех структурных факторов. Эти знаки непосредственно используются для предварительного построения рядов электронной плотности, из которых устанавливают положения других атомов. Включением этих других атомов в следующий набор вычисленных структурных факторов можно проверить, не окажутся ли некоторые из фаз на самом деле противоположными тем, которые были найдены при рассмотрении только тяжелого атома. [9]
При анализе структуры малых молекул первичную карту электронной плотности получают при помощи прямых методов определения структуры или, например, методом Паттерсона, устанавливая положение нескольких наиболее тяжелых атомов в молекуле. Такая первичная карта Фурье обычно недостаточно ясна и может указывать максимумы электронной плотности только для некоторых атомов в молекуле. Положение этих атомов определяется при интерпретации первичной карты и используется для расчета набора фаз. Этот набор затем применяется для расчета карты электронной плотности с помощью наблюдаемых структурных амплитуд, в результате чего уточняются положения других атомов. Вклад этих атомов может быть затем использован для расчета улучшенного набора фаз, что приводит к более совершенной карте электронной плотности. Подобная процедура последовательно повторяется до тех пор, пока не будут определены все атомные параметры. Для уточнения фаз и расчета карт электронной плотности могут быть использованы альтернативные циклы. Поскольку в случае малых молекул число экспериментально наблюдаемых параметров ( структурных амплитуд) велико по сравнению с числом переменных параметров, структура малых молекул определяется с высокой точностью. [10]
В молекулах с неаддитивными связями само понятие дипольного момента связи как независимой характеристики свойств отдельных связей теряет свой смысл. В этом случае параметры ik будут зависеть не только от свойств своих связей, но и от влияния окружения. Эта зависимость будет тем большей, и, следовательно, параметры все в меньшей степени будут отражать свойства отдельных связей, чем менее обособленными являются структурные элементы молекулы. В молекулах, состоящих из аддитивных групп, параметры каждой группы будут определяться только свойствами соответствующей группы и, в частности, будут являться функциями только таких естественных колебательных координат из совокупности всех естественных координат молекулы, которые определяют колебания этих групп как свободных молекул. В неаддитивных молекулах параметры Hk являются уже функциями не только межатомного расстояния своих связей, но зависят также от положения других атомов молекулы. [11]
В молекулах с неаддитивными связями само понятие дипольного момента связи как независимой характеристики свойств отдельных связей теряет свой смысл. В этом случае параметры [ ik будут зависеть не только от свойств своих связей, но и от влияния окружения. Эта зависимость будет тем большей, и, следовательно, параметры все в меньшей степени будут отражать свойства отдельных связей, чем менее обособленными являются структурные элементы молекулы. В молекулах, состоящих из аддитивных групп, параметры каждой группы будут определяться только свойствами соответствующей группы и, в частности, будут являться функциями только таких естественных колебательных координат из совокупности всех естественных координат молекулы, которые определяют колебания этих групп как свободных молекул. В неаддитивных молекулах параметры р / г являются уже функциями не только межатомного расстояния своих связей, но зависят также от положения других атомов молекулы. [12]
Авторов поразил тот факт, что из 10 отдельных энергетических минимумов ниже - 20 ккал / моль только 4 ассоциируются с молекулами воды, положение которых установлено кристаллографически. Аналогичным образом многие минимумы с энергией от - 15 до - 20 ккал / моль не имеют поблизости от себя молекул воды, входящих в кристалл. Большинство этих минимумов находится вблизи концов боковых цепей, состоящих из заряженных и других гидрофильных аминокислот, которые, как можно ожидать, специфически гидратированы. Из результатов уточнения кристаллографической структуры [5] с очевидностью следует вывод об интенсивном тепловом движении и / или разупо-рядочении длинных гидрофильных боковых цепей. Концевые атомы длинных боковых цепей характеризуются высоким значением температурного фактора, - и если даже их положение еще не установлено кристаллографическими методами, то им приписывают положения, согласующиеся с положениями других атомов и с классической стереохимией. В общем молекулы воды, связанные с этими боковыми цепями, подвержены еще более интенсивному тепловому движению и поэтому не могут давать легко идентифицируемых максимумов электронной плотности. [13]
О - Н не ориентированы постоянно в направлении одних и тех же соседних молекул. Выше 200 К диэлектрическая постоянная льда имеет величину такого же порядка, как и у воды; это показывает, что молекулы в электрическом поле могут очень легко изменять свою ориентацию. Кроме того, можно вычислить ( в хорошем согласии с экспериментом) величину остаточной энтропии льда, если предположить, что из четырех атомов водорода, окружающих каждый атом кислорода, два атома находятся ближе к данному атому кислорода, чем два другие атома водорода, и приблизительно направлены к двум из тех четырех атомов кислорода, которые тетраэдрически окружают данный атом кислорода. При этом эти два атома водорода не обязательно всегда направлены к одним и тем же двум атомам кислорода, и ориентация их может изменяться. В соответствии с этим представлением кристалл льда может принимать любую из большого числа конфигураций, отличающихся различным расположением атомов водорода в результате вращения некоторых молекул воды или перемещения некоторых атомов водорода из положений на расстоянии 1 А от одного атома кислорода в такое же положение вблизи другого атома кислорода данной связи. [14]
Страницы: 1