Постоянство - скорость - реакция
Cтраница 1
Постоянство скорости реакции при переходе от одного катализатора к другому вытекает из уравнений справедливых для реакции на равномерно-неоднородной поверхности. Если поверхность катализатора характеризуется экспоненциальным распределением, то из уравнения VII. [1]
Это постоянство скорости реакции не должно было бы иметь места, если бы первичными активными центрами являлись атомы О. [2]
Предположение о постоянстве скорости реакции справедливо, если изменения концентрации в реакторе невелики. [3]
В противном случае трудно было бы объяснить сохранение столь высокого постоянства скорости реакции и состава получаемых продуктов. [4]
Известный интерес представляет вопрос о том, сохраняется ли постоянство скоростей реакций, происходящих в полиэтилене, с увеличением дозы, как это обычно принято считать, или же скорость некоторых или всех реакций при более высоких дозах изменяется. При предельно больших дозах, когда общее число реакционноспособных групп значительно уменьшается, следует ожидать уменьшения скоростей всех реакций. Но возможно также, что изменение скоростей может происходить задолго до этой области доз, например, за счет исчерпывания определенных типов групп, особенно благоприятных для протекания данного типа реакций. Дол и его сотрудники [25, 31] считают, что образование водорода и двойных связей является линейной функцией дозы; Лоутон и его сотрудники [30] также наблюдали, что образование водорода идет пропорционально дозе до 200 мегафэр, но что образование гранс-виниленовых двойных связей при больших дозах идет медленнее. По-видимому, равновесие реакций образования водорода может смещаться при увеличении дозы, но таким образом, что общая скорость образования водорода остается постоянной. [5]
Равномерность облучения обеспечивает, при прочих равных условиях, постоянство скорости реакции хлорирования. [6]
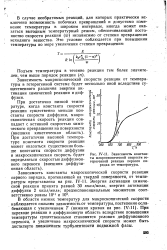 |
Зависимость константы макроскопической скорости гетерогенной реакции первого порядка от температуры. [7] |
В случае необратимых реакций, для которых практически исключена возможность побочных превращений и допустимо изменение температуры в широком интервале, иногда может оказаться выгодным температурный режим, обеспечивающий постоянство скорости реакции ( w) независимо от степени превращения исходного вещества. [8]
Некоторые отклонения в скорости реакции следует объяснить колебаниями температурного режима. Постоянство скорости реакции находится в связи с сохранением состава продуктов реакции. [9]
С точки зрения перекисной теории, постулирующей неизменяемость и постоянное присутствие катализатора, реакция окисления должна была бы в этот момент прекратиться. Следует отметить, что только часть низкомолекулярных органических солей марганца, осевших на стенки окислительной колонки, вообще говоря, может функционировать в качестве катализатора. Это и понятно, так как активная поверхность такого гетерогенного катализатора, по сравнению с поверхностью коллоидных частиц квазигетерогенных систем, чрезвычайно мала и поэтому вряд ли может оказать какое-либо влияние на ход реакции. Таким образом постоянство скорости реакции окисления и ее направления логически не согласуется с уменьшением активной поверхности катализатора, особенно, если последний действительно является переносчиком кислорода в течение всего времени процесса окисления. [10]
В первоначальный период исследований было указано, что при введении в реакционную зону крекинга олефинов или окиси азота скорость термического крекинга парафиновых углеводородов снижается до всегда одинакового предельного значения. При этом состав продуктов реакции не меняется. Это дало основание Хиншельвуду [232] считать, что ингибитор подавляет цепные реакции распада. Такая гипотеза хорошо объясняет постоянство скорости реакции при использовании различных ингибиторов. Однако она не может объяснить, почему продукты одного и того же состава получаются при крекинге с различным механизмом. [11]
До сего времени предполагалось, что скорость реакции со временем не меняется. Хотя это предположение, вообще говоря, неверно, во многих случаях оно существенно не влияет на результаты вычислений. Показано, что к моменту стремительного роста температуры в системе или к моменту взрыва успевает прореагировать лишь 1 % исходного вещества. Этим и оправдывается предположение о постоянстве скорости реакции до-взрыва. [12]
Так, в процессе цепного фотохимического хлорирования алканов ( в приведенном примере метана) образуется смесь хлорзамещенных продуктов. Обычно при фотохимическом хлорировании выход полихлорзамещенных выше по сравнению с другими способами хлорирования алканов. Равномерность облучения обеспечивает при прочих равных условиях постоянство скорости реакции хлорирования. [13]
При исследовании процессов жидкофазного окисления фенола нами было показано. Не исключено, что аналогичное явление имеет место и в случае окисления нефтепродуктов. При этом в фазе-смол оказываются наиболее реакционноспособные радикалы, что и приводит к торможению процесса. Это явление является определяющим на второй стадии процесса, когда образование смоляных ассоциатов идет преимущественно за счет наиболее активных радикалов, а в объем вытесняются менее-активные. В пользу этого также свидетельствует постоянство скоростей реакций расхода масел внутри всех трех стадий. [14]
Страницы: 1