Применение - корреляционное уравнение
Cтраница 1
Применение корреляционных уравнений в ряду полициклических соединений осложняется множественностью вариантов взаимного расположения заместителя и реакционного центра. Уже в случае нафталина даже при исключении из рассмотрения орто - и лери-положений каждому заместителю соответствуют два ряда констант по пять вариантов в каждом ряду, так как реакционный центр может находиться либо в а -, либо р-положе-нии. [1]
Применение корреляционных уравнений в ряду полициклических соединений осложняется множественностью вариантов взаимного расположения заместителя и реакционного центра. Уже в случае нафталина даже при исключении из рассмотрения орто - и леры-положений каждому заместителю соответствует два ряда констант по пять вариантов в каждом, так как реакционный центр может находиться либо в а -, либо в р-положении. Для описания нафталиновых констант заместителей ац необходимо указание двух индексов: t - - для положения заместителя в ядре и / - для положения реакционного центра. [2]
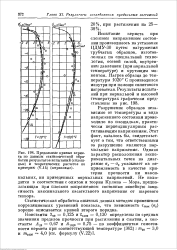 |
Предельные кривые кермета до данным статистической обработки результатов испытаний ( сплошные и теоретических расчетов по критерию ( штриховые. [3] |
Статистическая обработка опытных данных методом применения корреляционных уравнений показала, что зависимость ттах ( о о) хорошо описывается кривой второго порядка. [4]
Более подробно о рекомендуемых практических правилах применения корреляционных уравнений см. статью В. А. Пальма в сб. [5]
В блоке реактора 3 превышены границы применения корреляционных уравнений. Для продолжения расчетов взято экстраполированное значение. [6]
Нами уже были упомянуты некоторые работы по применению корреляционных уравнений для изучения ради-кал Шых реакций [192 - 194], выполненные в конце 40 - начале 50 - х годов. Теперь рассмотрим их несколько подробнее. [7]
Большой теоретический и практический интерес представляют исследования по применению корреляционных уравнений к константам диссоциации производных фосфорных и фосфиновых кислот. [8]
В двух последних случаях было найдено сильное смещение равновесия соответственно в тионовую и лактам-ную формы. Интересные возможности количественного метода были вскрыты при применении корреляционных уравнений. Кабачником количественная теория прототропного таутомерного равновесия позволила объяснить большое число экспериментальных фактов и значительно продвинуться в изучении разнообразных таутомерных систем. Проблемы таутомерии включают не только равновесие, но и механизм таутомерных превращений. Вопросы механизма таутомерных равновесий были в основном разработаны в Англии. [9]
В настоящее время в проблеме связи между химическим строением веществ и их реакционноспособностью существует много пока еще не ясных вопросов. Для решения этой проблемы сейчас намечаются два основных пути. Первый путь - это применение корреляционных уравнений Гаммета - Тафта, позволяющих количественно выражать относительное влияние тех или иных заместителей на термодинамические или кинетические константы реагирующих молекул, принимая влияние какого-то определенного заместителя за стандартное. Второй путь связан с попытками кванто-во-химического расчета распределения плотности электронов в реагирующих молекулах. [10]
Познакомимся более подробно с предпосылками, лежащими в основе применения корреляционных уравнений. [11]
Используемые при этом методы всегда требуют привлечения исходных экспериментальных данных, а применяемые теоретические представления неизбежно будут в какой-то степени интуитивными, полуэмпирическими и не всегда могут быть во всех деталях строго обоснованными. Однако простота применения этих методов и большая универсальность, позволяющая совместно рассматривать широкий круг реакций и объектов, делают их очень важными и полезными как для трактовки реакционной способности органических соединений, так и для изучения механизмов органических реакций. В настоящее время наиболее широко используются два количественных подхода к реакционной способности органических соединений. Первый из них - применение корреляционных уравнений и корреляционного анализа - существует уже более сорока лет. Второй - использование приближенных квантовомеханических моделей, основанных на применении теории возмущений молекулярных орбиталей, - начал интенсивно развиваться только в последние годы, но находит все более широкое применение. В отличие от строгих квантовомеханических расчетов, приложимых в основном к отдельным молекулам и не дающих возможности использовать сравнение реакционной способности структурно близких соединений, метод возмущений молекулярных орбиталей плодотворно применяется для общей трактовки реакционной способности органических молекул. Подробное рассмотрение этих концепций выходит за рамки этой книги и привело бы к недопустимому росту ее объема. Поэтому далее будут рассмотрены только основы и простейшие приложения этих методов. Для более глубокого знакомства с ними следует обратиться к специальной литературе. [12]
Страницы: 1