Газо-жидкостный вариант
Cтраница 1
Успех газо-жидкостного варианта, связанный с исключительной однородностью жидкости и с возможностью сравнительно легкого регулирования свойств этой жидкости, был, однако, неполным, ибо недостатки этого варианта хроматографии оставались не устраненными. [1]
При газо-жидкостном варианте хроматографии в результате повышения температуры колонки увеличивается упругость пара стационарной фазы, что вызывает дрейф нулевой линии и уменьшает срок службы колонки. Все ото приводит к необходимости находить способы, если не совсем ликвидировать дрейф, то во всяком случае уменьшить его до приемлемой величины. Одной, по-видимому, наиболее действенной мерой является применение схемы со второй компенсирующей колонкой, принцип действия которой был изображен на рис. 25, в. При такой схеме рабочая и вспомогательная ( компенсирующая) колонки находятся в одинаковых температурных условиях, и таким образом, все изменения в потоке газа-носителя будут одинаковыми в обеих линиях - через сравнительную и измерительную камеры детектора. Преимуществом такой схемы, также способствующим устойчивости нулевой линии, является то, что в обеих камерах детектора давление газа-носителя одинаково ( соединены с атмосферой), что не наблюдается в обычных одноколоночных схемах, где в сравнительных камерах, находящихся перед колонкой, всегда давление выше, чем в измерительной. [2]
Насадки для газо-жидкостного варианта, который в основной используется в настоящее время в препаративной хроматографии, не могут полностью удовлетворить эти требования, так как применяемые неподвижные жидкие фазы ( НЖФ), как правило, дороги, а производство их в больших количертвах иногда затруднительно. Кроме того, наличие в колоннах жидкой фазы накладывает определенные температурные ограничения. При длительном использовании таких колонн для разделения больших доз происходит унос неподвижной фазы из насадки, сорбент теряет свою разделяющую способность и не регенерируется, а отбираемые фракции чистых веществ загрязняются парами НЖФ или продуктами их деструкции. [3]
В случае газо-жидкостного варианта селективной фазой является пленка малолетучей жидкости, нанесенная на поверхность инертного носителя. Одним из лучших носителей для газо-жидкостной хроматографии является стекло [2] благодаря инертности, чистоте и однородности поверхности. Недостатком стекла является малая удельная поверхность, что требует применения небольших проб и высокочувствительных детекторов. Пористые стекла являются продуктом обработки щелочно-боросиликатных стекол растворами кислот и водой. При этом имеет место избирательная растворимость стекла. В раствор переходят практически только окислы щелочных металлов и В203, a Si02 остается в виде пористого нерастворимого остатка. Структура и характеристика полученного таким образом пористого стекла определяется как химической устойчивостью исходного материала, так и пространственным распределением В203 и окислов щелочных металлов в кремнеземистом остове. [4]
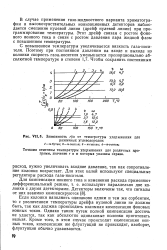 |
Зависимость л / со от температуры удерживания для различных углеводородов. [5] |
В случае применения газо-жидкостного варианта хроматографии и высокочувствительных ионизационных детекторов наблюдается смещение нулевой линии ( дрейф нулевой линии) при, программировании температуры. Этот дрейф связан с ростом фонового ионного тока в связи с ростом давления пара жидкой фазы с повышением температуры. [6]
Сводка литературных данных по хроматографическому разделению ароматических углеводородов в газо-жидкостном варианте за период 1960 - 1966 и, частично, 1967 гг. дана в таблице. В ней собран материал не только из работ, посвященных специально разработке аналитических методик, но также из сообщений, в которых вопросы хроматографии играли подсобную роль. В связи с постоянно растущим числом работ по применению хроматографических методов для анализа ароматических углеводородов представленная сводка литературных данных, возможно, не является исчерпывающей. [7]
Полученные данные показали, что величины удерживания молекул в газо-жидкостном варианте хроматографии при использовании в качестве твердого инертного носителя пористых полимерных сорбентов существенно зависят, с одной стороны, от природы и количества неподвижной жидкой фазы, а с другой - от особенностей структуры и химии поверхности полимерного сорбента. [8]
Анализ галогенидов металлов методом газо-адсорб-ционной хроматографии имеет определенные преимущества по сравнению с газо-жидкостным вариантом. [9]
Хотя уравнение ( 11 98) было выведено для жидкостно-жидко-сткой хроматографии, оно справедливо и для газо-жидкостного варианта. Следует иметь в виду, что результаты, полученные на колонке со смешанным сорбентом [ согласно уравнению ( 11 68) ], будут существенно отличаться от результатов, полученных на колонке со смешанной фазой, лишь тогда, когда значения Vgl и Vsz заметно отличаются друг от друга. [10]
В нашей работе проведено разделение и анализ смеси некоторых хлорированных фенолов с применением высокоэффективной капиллярной колонки в газо-жидкостном варианте. [11]
Основное внимание было сконцентрировано на разделении и анализе легких и углеводородных газов, сначала по газо-адсорбционному, несколько позднее по газо-жидкостному варианту, вначале фронтальным и вы-теснительным, а затем проявительным методом. При этом если вначале использовалась макроаналитическая шкала, то постепенно был совершен переход в область микро - и субмикрошкалы. Число публикаций по этим вопросам за последние 20 лет росло очень стремительно и в настоящее время достигает нескольких тысяч в год. [12]
Разработаны газохроматографические методы определения окиси углерода, сернистого и серного ангидридов и многих других соединений. В случае газо-жидкостного варианта хроматографии при использовании в качестве жидких фаз силиконовых масел или фторированных углеводородов получаются невоспроизводимые пики с сильно размытыми хвостами. В случае газоадсорбционного варианта использование в качестве сорбента силикагеля и активированного угля позволяет получить симметричные пики двуокиси азота, однако возникает трудность ее определения в присутствии двуокиси углерода вследствие их наложения на хроматограмме. [13]
Поэтому средняя концентрация силовых центров на поверхности даже плотного монослоя меньше, чем на поверхности твердого тела, следовательно, энергия неспецифического дисперсионного взаимодействия с поверхностью плотного монослоя невелика. Отсюда вытекают требования к адсорбенту - носителю монослоя. В отличие от носителей жидкости в газо-жидкостном варианте хроматографии, адсорбент - носитель монослоя должен быть сильно, но неспецифически адсорбирующим, геометрически достаточно однородным и должен обладать достаточно высокой удельной поверхностью, что необходимо для обеспечения нужной эффективности колонки, при меньшей энергии адсорбции. [14]
Смит и Вэддингтон [29] при проведении газохроматогра-фического анализа алифатических спиртов, диолов и эфиров на ароматических полимерах ПАР-1 и порапак Q отметили, что для отдельных классов соединений наблюдается линейная зависимость логарифма времени удерживания оттемпе-ратуры кипения элюента. Небольшие отклонения от линейности обнаружены при изменении структуры алкильной цепи. Характер удерживания спиртов, диолов, эфиров на исследованных полимерах оказался в основном аналогичным удерживанию этих соединений в газо-жидкостном варианте хроматографии на колонках, заполненных сорбентом целит-апиезон L, хотя времена удерживания на них гораздо меньше. [15]
Страницы: 1 2