Недостаточное разделение
Cтраница 1
 |
Кривые извлечения для нескольких разгонок. [1] |
Недостаточное разделение, наблюдающееся в случае однократной молекулярной дистилляции, означает, что при перегонке сложной смеси любой компонент будет иметь определенную концентрацию в той или иной фракции. Если фракции отбирают через равные промежутки времени при равномерном возрастании температуры, то изменение выхода компонента А в фракциях дистиллята следует кривой Гаусса. Кривые распределения концентраций каждого компонента перекрывают друг друга согласно схеме, показанной на рис. IX-40. Температура, соответствующая максимуму кривой, полученному в стандартных условиях, является характерной для молекулярной дистилляции величиной, заменяющей точку кипения. [2]
 |
Схема хроматографической колонки непрерывного. [3] |
При недостаточном разделении снова производят переключение газа-носителя в колонку / и так повторяют до полного разделения смеси. [4]
Изучить возможности количественного анализа в условиях недостаточного разделения компонентов смеси при использовании дифференциального выделения сигнала детектора, слабо искаженного наличием мешающего компонента. [5]
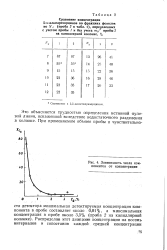 |
Зависимость числа компонентов от концентрации. [6] |
Это объясняется трудностью определения истинной нулевой линии, искаженной вследствие недостаточного разделения в колонке. [7]
Метод ДВС значительно ( примерно на порядок) уменьшает погрешность измерения времен удерживания, обусловленную недостаточным разделением хроматографических зон в колонке; отсчет производится по пересечению с осью абсцисс круто ниспадающей или круто восходящей ветви дифференциально выделенного сигнала, что приводит к меньшей инструментальной погрешности результата. При отсутствии интегрирующих устройств появляется возможность надежного измерения времен удерживания компонентов, пики которых на обычных хроматограммах оказываются зашкаленными. [8]
При термическом разложении тшлив и полимеров, протекающем частично по случайному распределению, как правило, образуются сложные смеси с широким диапазоном молекулярных масс. Анализ подобных омесей характеризуется большим числом разделяемых компонентов на единицу шкалы удерживания, вызывающий недостаточное разделение омесей, некоторую неопределенность нулевой линии ( особенно в ус - ЛО1ВИЯК программирования температуры), отсутствием данных чувствительности дифференциального детектора к отдельным компонентам пробы, а также возможностью необратимого удерживания части пробы в хроматографической аппаратуре. При синтезе непосредственно до анализа менее полярных производных анализируемых соединений положение усложняется необходимостью учета стехиометрии соответствующей реакции. [9]
Пятна, образованные неизвестными соединениями. Неидентифицированы из-за недостаточного разделения. [10]
Вернемся к примерам, представленным на рис. 111.31. На хроматограммах д - и все вещества имеют хорошее удерживание, однако удовлетворительными можно считать только хроматограм-мы д, ж, и. На хроматограммах е и з отдельные пары пиков имеют недостаточное разделение. В таких случаях мы говорим, что селективность системы недостаточна. Селективность системы, в которой используется данная неподвижная фаза, в первую очередь определяется селективностью используемой подвижной фазы. Селективность подвижных фаз связана с их способностью к специфическим взаимодействиям с сорбатамн, обладающими определенными структурными признаками. Она проявляется в том, что точное значение элюирующей силы данного растворителя или смеси по отношению к сорбатам различного строения может быть различным. Селективность, как и элюирующая сила бинарной подвижной фазы, определяется в первую очередь природой более сильного ее компонента. [11]
Вернемся к примерам на рис. 3.1. На хроматограммах д-и у всех веществ наблюдается хорошее удерживание, однако удовлетворительными можно считать только хроматограммы д, ж, и. На хроматограммах е и з отдельные пары пиков имеют недостаточное разделение. В таких случаях говорим, что селективность системы недостаточна. Селективность системы, в которой используется данная неподвижная фаза, в первую очередь определяется селективностью используемой подвижной фазы. Селективность последней связана с ее способностью к специфическим взаимодействиям с сорбатами, обладающими определенными признаками. Она проявляется в том, что точное значение элюирующей силы данного растворителя или смеси по отношению к сорбатам различного строения может быть различным. Селективность, как и элюирующая сила бинарной подвижной фазы, определяется прежде всего природой более сильного ее компонента. [12]
Вернемся к примерам, представленным на рис. 111.31. На хроматограммах д - и все вещества имеют хорошее удерживание, однако удовлетворительными можно считать только хроматограм-мы д, ж, и. На хроматограммах е и з отдельные пары пиков имеют недостаточное разделение. В таких случаях мы говорим, что селективность системы недостаточна. Селективность системы, в которой используется данная неподвижная фаза, в первую очередь определяется селективностью используемой подвижной фазы. Селективность подвижных фаз связана с их способностью к специфическим взаимодействиям с сорбатамн, обладающими определенными структурными признаками. Она проявляется в том, что точное значение элюирующей силы данного растворителя или смеси по отношению к сорбатам различного строения может быть различным. Селективность, как и элюирующая сила бинарной подвижной фазы, определяется в первую очередь природой более сильного ее компонента. [13]
Вернемся к примерам на рис. 3.1. На хроматограммах д-и у всех веществ наблюдается хорошее удерживание, однако удовлетворительными можно считать только хроматограммы д, ж, и. На хроматограммах е и з отдельные пары пиков имеют недостаточное разделение. В таких случаях говорим, что селективность системы недостаточна. Селективность системы, в которой используется данная неподвижная фаза, в первую очередь определяется селективностью используемой подвижной фазы. Селективность последней связана с ее способностью к специфическим взаимодействиям с сорбатами, обладающими определенными признаками. Она проявляется в том, что точное значение элюирующей силы данного растворителя или смеси по отношению к сорбатам различного строения может быть различным. Селективность, как и элюирующая сила бинарной подвижной фазы, определяется прежде всего природой более сильного ее компонента. [14]
Абсолютные значения погрешностей, приведенные в работе [106], относятся к случаю малокомпонентных искусственных смесей и хорошо разделенных пиков. Для реальных смесей значения погрешностей оказываются более высокими из-за асимметрии пиков, присутствия примесей, недостаточного разделения и других причин. Авторы не анализируют методы с точки зрения точности при равной затрате труда. При таком подходе анализ методом внутреннего стандарта нужно сопоставлять с методом стандартной добавки с учетом того факта, что первый из этих методов требует дополнительного многократного хроматографирования для нахождения относительного калибровочного коэффициента. Очевидно, что усреднение даньгх для нескольких добавок увеличит точность метода стандартной добавки. [15]
Страницы: 1 2