Калориметрическая величина
Cтраница 1
Калориметрические величины представляют интегральные теплоты адсорбции для моля пара, адсорбированного на 500 г угля ( 44 8 с. Изостерические теплоты были вычислены согласно уравнению ( 24) из графиков 1пр относительно ijT для постоянных количеств адсорбированного пара. За исключением четыреххлористого углерода, графики дают хорошие прямые линии с приблизительно одинаковыми наклонами; это указывает на то, что дифференциальные теплоты адсорбции не сильно изменяются с количеством адсорбированного пара. [1]
Отсюда видно, что корреляция калориметрических величин энтропии с рассчитанными по молекулярным постоянным может быть достигнута только при условии учета динамического взаимодействия волчков ( для диметилдихлорсилана поправка на взаимодействие волчков составляет - 0 3 энтр. Только при этих условиях величины тормозящих потенциалов оказываются вполне разумными и согласующимися с оценками, полученными из спектральных данных. [2]
В рассмотренном варианте метод низкотемпературной калориметрии использован для определения калориметрических величин, необходимых для вычисления констант химической термодинамики - стандартных энтропии и энтальпий значительного ряда ллкнл - и арилхлорсиланов. [3]
Мы провели также расчеты для некоторых двойных систем, для которых данные о равновесии жидкость-пар и калориметрические величины теплот смешения были получены в нашей лаборатории. На рис. 4 изображены кривые теплот смешения для систем н-пропиловый спирт-вода и пропилацетат - - про-пиловый спирт, вычисленные из данных о равновесии жидкость-пар и измеренные калориметрически. [4]
Этот вывод подтверждается сравнением результатов калориметрических и спектральных экспериментов. Калориметрическая величина, обычно и называемая в химической литературе энергией связи, не есть, таким образом, локальная характеристика только данной химической связи, а зависит ( иногда довольно сильно, например, для связей в циклических соединениях) от поведения всего окружения. [5]
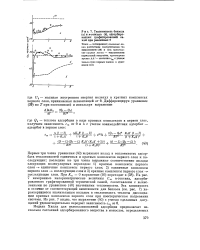
Первые три члена уравнения ( 40) выражают вклад в теплоемкость адсор-бата теплоемкостей единичных и кратных комплексов первого слоя и последующих; последние же три члена выражают соответственно вклады следующих молекулярных переходов: 1) кратные комплексы первого слоя - единичные комплексы первого слоя; 2) единичные комплексы первого слоя - последующие слои и 3) кратные комплексы первого слоя - последующие слои. На рис. 7 измеренные калориметрические величины Ст к-гексана, адсорбированного графитированной термической сажей, сопоставлены с вычисленными по уравнению ( 40) значениями теплоемкости. Эта зависимость в отличие от соответствующей зависимости для бензола ( см. рис. 7) характеризуется значительным вкладом в теплоемкость теплоты диссоциации кратных комплексов первого слоя при изостерическом нагревании системы. [7]
Первые три члена уравнения ( 40) выражают вклад в теплоемкость адсор-бата теплоемкостей единичных и кратных комплексов первого слоя и последующих; последние же три члена выражают соответственно вклады следующих молекулярных переходов: 1) кратные комплексы первого слоя - единичные комплексы первого слоя; 2) единичные комплексы первого слоя - последующие слои и 3) кратные комплексы первого слоя - последующие слои. На рис. 7 измеренные калориметрические величины Ст к-гексана, адсорбированного графитированной термической сажей, сопоставлены с вычисленными по уравнению ( 40) значениями теплоемкости. Эта зависимость в отличие от соответствующей зависимости для бензола ( см. рис. 7) характеризуется значительным вкладом в теплоемкость теплоты диссоциации кратных комплексов первого слоя при изостерическом нагревании системы. [8]
Qd и qst в пределах погрешности эксперимента практически совпадают. Подобный пример для адсорбции воды на сульфате бария приведен на рис. XIV-24. Обращает внимание также совпадение изостерических и калориметрических величин на рис. XIV-25, в. Вообще говоря, вопрос о соответствии Калориметрических и термодинамических те плот адсорбции не так уж тривиален. [9]
В пределах каждой группы плавок наблюдается большой разброс величин подсчитанных термичностей. Значение термичности, подсчитанной исходя из полного веса металла, удовлетворительно совпадает с калориметрической; разница, составляющая 5 45 - 12 9 ккал, объясняется тем, что при калориметрическом опыте замеряются и теплоты неосновных процессов. Разброс подсчитанных значений термичностей меньше предыдущего случая и одинаков с разбросом калориметрических величин. Сопоставление значений термичностей, полученных из калориметрического опыта и из расчета, говорит о том, что рассчитанная термичность должна определяться, исходя только из полного веса металла. [10]
В целях удобства изложения вопрос о свободной энергии будет рас смотрен в следующих главах, а в этой главе мы рассмотрим те величины, которые могут быть получены из данных о свободной энергии, хотя такое расположение материала и может показаться несколько произвольным. Затем будет весьма подробно разобран вопрос о парциальной молярной теплоемкости, абсолютное значение которой может быть вычислено. Далее будет изложен также вопрос о теплоте нейтрализации, и таким образом все эти калориметрические величины можно будет рассматривать в непосредственной связи друг с другом. Последние параграфы будут посвящены парциальным молярным объемам, расширяемости и сжимаемости. [11]
В целях удобства изложения вопрос о свободной энергии будет рас смотрен в следующих главах, а в этой главе мы рассмотрим те величины, которые могут быть получены из данных о свободной энергии, хотя такое расположение материала и может показаться несколько произвольным. Затем будет весьма подробно разобран вопрос о парциальной молярной теплоемкости, абсолютное значение которой может быть вычислено. Далее будет изложен также вопрос о теплоте нейтрализации, и таким образом все эти калориметрические величины можно будет рассматривать и непосредственной связи друг с другом. Последние параграфы будут посвящены парциальным молярным объемам, расширяемости и сжимаемости. [12]
К настоящему времени экспериментальные данные о равновесии жидкость - пар при различных значениях температуры ( или давления) известны для нескольких сотен бинарных и нескольких десятков тройных систем. Как простота самого метода, так и сравнительная доступность исходных для расчета данных делают рассматриваемый метод определения те-плот смешения весьма привлекательным. Однако в литературе недостаточное внимание обращается на оценку точности получаемых расчетом величин теплот смешения, часто точность преувеличивается. Для того чтобы наглядно оценить существующее положение, мы провели сравнение опубликованных различными авторами данных о теплотах смешения жидкостей, полученных расчетом, с результатами калориметрических измерений, которые можно принять за истинные. На диаграммах калориметрическим величинам теплот смешения соответствуют сплошные линии, рассчитанным из данных о равновесии жидкость - - пар - пунктирные. Как видно из рисунка, из девяти систем только для трех ( системы С6Н6 - СС14, С6Н12 - СС14 и - C7Hi6 - C6Hi2) имеет место вполне удовлетворительное согласование данных, для некоторых из остальных систем расхождения весьма значительны. [13]
VII, 3) и ( VII, 4) в общем случае непригодны. Иными словами, нельзя точно указать, к какой температуре относится найденная по уравнениям или из наклона на прямой lgP s ( T-1) ( рис. 45) величина ДЯпар. При температурах, далеких от критической, числитель и знаменатель уравнения ( VII, 2) оказываются примерно одинаково преувеличенными, вследствие чего неверные по существу допущения не сказываются на левой части этого уравнения. VII, 2 - 4), можно считать практически равной ( ДЯ ар. Однако в силу допущений, сделанных при выводе уравнения ( VII, 4), вычисленные по этому уравнению значения теплоты парообразования даже при Р - 1 несколько превышают калориметрические величины. [14]
Страницы: 1