Водный раствор - хлористый барий
Cтраница 2
Результаты экспериментов позволяют рекомендовать следующий метод получения титаната бария. Готовятся одномолярные водные растворы хлористого бария и четыреххлористого титана. Полученные растворы анализируются на содержание бария и титана, после чего смешиваются в строго эквимолярных отношениях. Осадитель готовится смешиванием 1 л 2.3 - 2.5 мол. Оба приготовленных раствора сливаются параллельно из двух сосудов в реакционную емкость. В производственных условиях этот процесс может быть легко автоматизирован. Полученные описанным выше способом осадки отжимаются под вакуумом или на центрифуге и отмываются водой от хлорид-ионов. Такие осадки помещаются в холодные камерные печи с силитовыми нагревателями и медленно, со скоростью 150 - 200 град. Такой режим термического разложения обеспечивает получение рассыпчатого мелкодисперсного порошка титаната бария. [16]
 |
Стружкоотделительные канавки на задней поверхности сверла. [17] |
Для жаропрочных сплавов рекомендуется 50 % - ная эмульсия или водный раствор хлористого бария с добавкой 1 % - ного нитрата натрия, для титановых сплавов - касторовое и осерненное масла, олеиновая кислота или ее смеси. [18]
Щелочные соли худо кристаллизуются, водные растворы их дают с водным раствором хлористого бария в холоде осадок, подобный клейстеру, липкий; в горячей воде осадок спекается или свертывается, как творог, но почти вовсе не растворяется; в смеси двух объемов 95 % спирта с одним объемом воды осадок этот растворяется легко ( но труднее соответствующей соли этиламаровой кислоты), при кипячении и при охлаждении выделяется в виде мелких, микроскопических иголок, срастающихся в пучки или в шарики. Чистая, два раза перекристаллизованная баритовая соль содержала 15 03 % бария, 0 352 хорошо высушенной при 110 С соли дали 0 090 сернокислого барита; формула С5оН4б04Н - Н6Ва04 требует 14 9 % бария. [19]
Поверхность изделия тщательно очищают от загрязнений и обрабатывают в специальных растворах. Перед элиминированием изделия подвергают офлюсовке в горячем 2 % - ном водном растворе хлористого бария и хлористого натрия, взятых в соотношении 70: 30 и выдерживают 5 - 10 мин или производят пассивацию в 1 - 3 % - ном растворе хромового ангидрида при температуре 18 - 20 в течение 0 5 - 1 мин. Перед операцией цинковые изделия подвергают флюсованию в хлористом аммонии и хлористом цинке. [20]
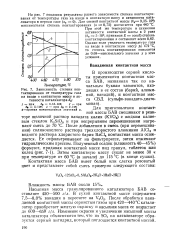 |
Зависимость степени контактирования от температуры газа на входе в контактную массу и активности катализатора. 0. [21] |
Для приготовления контактной массы БАВ смешивают в реакторе щелочной раствор ванадата калия ( KVO3) с жидким калиевым стеклом K2SiO3 и при непрерывном перемешивании нагревают смесь до 70 С. После добавления в смесь ( при перемешивании) солянокислого раствора треххлористого алюминия А1С13 и водного раствора хлористого бария ВаС12 контактная масса осаждается. Ее отфильтровывают на фильтрпрессе, затем отжимают гидравлическим прессом. [22]
Приготовляя процентный раствор из сухой соли, нельзя во избежание неточности прибавлять навеску к заданному объему жидкости. Следует растворить ее сначала в меньшем объеме жидкости, а затем перелить раствор через фильтр в мерную колбу нужного объема и довести его до черты растворителем. Например, если необходимо приготовить 10 % - шли водный раствор хлористого бария, то нельзя 10 г ВаС12 - 2Н20 добавлять к 100 мл дистиллированной воды, а надо растворить навеску соли в50 - 60 мл воды в химическом стаканчике и профильтровать раствор в мерную колбочку емкостью 100 мл. Многократно споласкивая, схакашиш, ттоъвм колбочки доводят промывной водой через фильтр до черты. [23]
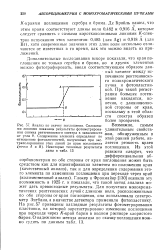
Глокер и фролмайар [130] оценили эту возможность в 1925 г. и показали, что такой метод анализа может дать превосходные результаты. На рис. 57 приведены наглядные результаты фотометрирования пластинки, на которой зарегистрировано изменение поглощения при переходе через / ( - рай бария в водном растворе хлористого бария. [25]
Однако при анализе производственных продуктов определения а-сульфокарбоновых кислот мешают примеси других Сульфопроизвод-ных, которые титруются вместе с сульфогруппой сульфокарб. Поэтому по полученной кривой титрования определяют лишь содержание серной кислоты. Для йпределения а-сульфокарбоновых и жирных кислот предполагается проводить титрование второй аликвотной части в присутствии соли бария. Это титрование проводят аналогична первому, до появления двух скачков потенциалов, после чего прибавляют избыток водного раствора хлористого бария, и продолжают титрование до появления нового скачка потенциала. [26]
Страницы: 1 2