Состояние - реакция
Cтраница 1
Равнозесное состояние реакции ( 4 - 1) может быть, как уже указывалось, достигнуто с обеих сторон, что было доказано Mayer и Altmayer [13], которые исследовали равновесные соотношения в системе метан-водород-углерод при давлении 750 мм. С; Pring и Fair-lie [14] продолжили эти исследования, расширив интервал температур и давлений, и нашли что давление благоприятно влияет на образование метана из угля и водорода, что вполне понятно, так как реакция идет с уменьшением объема. [1]
Такое состояние реакции называется переходным состоянием. Если принять k2 ki, то k2 - ki yk2 и соотношение e - klt erkJ достигается при меньших временах реакции. [2]
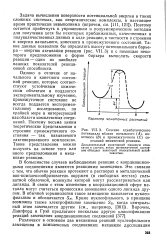 |
Сечение адиабатического потенциала вблизи начального ( А, конечного ( В и промежуточного ( С состояний химической реакции. [3] |
Однако в отличие от начального и конечного состояний реакции, которые соответствуют устойчивым химическим объектам и поддаются экспериментальному изучению, промежуточное состояние не всегда поддается экспериментальному исследованию, по крайней мере в интересующей нас области комплексных соединений. [4]
То же самое верно для трех фиксированных групп в переход-лом состоянии реакции 5Л2 ( см. стр. В трех - и четырехчленных ( малых) циклах истинные валентные углы близки к ВО и 90 соответственно. При наличии у этих атомов двойных связей ( карбонильные соединения): ти последние будут очень легко вступать в реакции присоединения, В пяти-членных циклах имеет место другая ситуация, обусловленная иными причинами. В цпклопентане с его тетраудрическнм углеродом осуществляются десять взаимодействий противостоящих друг другу ( ] - Н - связей, что приводит к значительному конформационному напряжению ( см. гл. [5]
Классификация реакций замещения на нуклеофильные и электрофильные дает только картину начального и конечного состояний реакции. Общее изменение системы не определяется этими граничными состояниями, а связано главным образом с механизмом реакции. Поэтому реакции классифицируют по числу частиц ( ионов или радикалов), взаимодействие которых приводит к химическому превращению; это определяет молеку-лярность реакции. Молекулярность реакции всегда отражает стадию, определяющую общую скорость реакции, и соответствует числу молекул, в которых на этой стадии происходит изменение числа ковалентных связей. [6]
Сравнение состояния сегрегации с уровнем молекулярного смешения для эндотермических реакций показывает, что наивысшая степень превращения достигается при сегрегированном состоянии реакций всех порядков. Разница между этим результатом и результатом, рассмотренным при изотермических условиях, для которых существен порядок реакции, обязана характеру изменения - скорости процесса. В эндотермической системе скорость уменьшается с увеличением степени превращения вследствие расходования реагентов и уменьшения температуры системы. Для описанных выше систем температурный эффект был большим, чем компенсация, обусловленная порядком реакции. [7]
Таким образом, принцип смещения равновесий Ле-Шателье позволяет заранее предсказывать, в какую сторону сместится равновесие при изменении параметров состояния реакции, и наоборот, какое изменение следует внести в условия проведения процесса для увеличения выхода продукта. Однако для количественных нового положения равновесия и теоретического протекания реакции необходимы расчеты с использованием соответствующих уравнений в интегральной форме. [8]
В общем случае, однако, замораживание равновесий не обязательно должно быть связано с сильным различием энтропии начального и конечного состояний реакции. К подобным эффектам может приводить изменение дипольного момента и, следовательно, энтропии системы в промежуточных областях координаты реакции, в частности в области активационного барьера. Можно, вообще говоря, представить себе и такую ситуацию, когда дипольный момент комплекса или молекулы в ходе реакции не меняется ( или почти не меняется) по величине, но зато меняется его направление. Энтропия системы в этом случае не меняется, однако возможность переориентации молекул растворителя зависит от вязкости среды, и равновесие может заморозиться. [9]
 |
Линейная связь свободной. [10] |
Поэтому существование этих соотношений должно означать, что переменный вращательный фактор для кислот формулы ХСН2СООН почти совершенно гасится при переходе от соединений, взятых в исходный момент реакции к переходному ( или конечному) состоянию реакции. Именно такой тип погашения факторов, связанных с кинетическими энергиями движений, наблюдается в относительных константах скорости [ для реакций, подчиняющихся уравнениям ( 7) и ( 16) ], причем они являются, по существу, количественной мерой полярных влияний. [11]
Основные выводы, к которым пришел автор при рассмотрении различных экспериментальных данных, следующие: отношение KOTS / нвг действительно может быть использовано в ряде случаев как мера степени разрыва связи С-X в переходной состоянии реакций замещения и отщепления. Отношение тозилат / бро-вдд является маленьким ( имеет значение около единицы), если в реакции участвует сильный нуклеофил и если субстрат имеет слабую тенденцию к ионизации. Такая картина наблюдается для реакций, где, например, в качестве нуклеофила используются тиолат-ионы в различных растворителях или галогенид-ионы в апротонной среде, а также для реакций ( в том числе реакций элиминирования оЕ 1сВ - характером), которые включают в себя значительное образование связи. Обычно это отношение мало, если используются такие субстраты, как метил - или первичные алкилпроизводные. [12]
Пока обсуждаются только первичные пространственные эффекты, которые непосредственно зависят от определенных различий в энергиях отталкивания. Если существует различие между первоначальным и переходным состояниями реакции, то наблюдаются кинетические пространственные эффекты. Наиболее важной группой подобных эффектов является та, в которой энергия отталкивания в переходном состоянии больше, чем в исходном. Сюда относятся кинетические эффекты, объединяемые термином пространственные затруднения, который естественно сохранен по соображениям историческим, хотя было бы удобнее заменить его более точным термином пространственное замедление, чтобы, таким образом, было ясно, что речь идет о затруднении кинетическом, а не термодинамическом. Следует полагать, что в некоторых реакциях, особенно в тех, которые зависят от скорости гетеролиза, определяющего скорость реакции, энергия отталкивания в переходном состоянии меньше, чем в исходном. В этом случае возможен второй тип кинетического пространственного эффекта, а именно пространственное ускорение. [13]
Как известно, методы современной квантовой химии не позволяют теоретически вычислять абсолютные значения энергии активации химических реакций с точностью, которая была бы достаточна для решения вопросов кинетики и механизма химических реакций. Однако квантовая химия, используя идею о пере ч) дном состоянии реакции, позволяет теоретически обсудить вопрос о влиянии различных факторов на величину энергии активации в ряду однотипных реакций. Несколько лет назад автором [ 2, 4j был предложен другой метод рассмотрения этого вопроса. [14]
В следующих разделах количественно рассмотрены термодинамика и кинетика установления равновесия и влияние различных структурных особенностей на состав смеси гомологов при равновесии. Однако при термодинамическом рассмотрении мы имеем дело только с начальным и конечным состояниями реакции и с различием между свободными энергиями реагентов и продуктов. Вопрос о том, как протекает полимеризация или деполимеризация, термодинамика не рассматривает. [15]
Страницы: 1 2