Последующая стадия - превращение
Cтраница 1
Последующие стадии превращения бензоилсалицилата меди определяются природой среды. В протонном растворителе ( например, вода или бензойная кислота) бензоилсалицилат меди декарбоксилируется с образованием фенилбензоата, дающего при гидролизе фенол. В апротонных ароматических растворителях ( бензол и его гомологи) в основном образуется фенилбензоат и в небольшом количестве - сложные эфи-ры бензойной кислоты и растворителя. В связи с этим было высказано предположение о возможности другой пространственной конфигурации бензоата меди, способствующей межмолекулярному взаимодействию бензоата меди с молекулами растворителя. [1]
На последующих стадиях превращения зостерового материала ( в песках) 2li31 отличие битумов от исходного органического вещества выступает резче и соответствующая точка на треугольнике ( обр. [2]
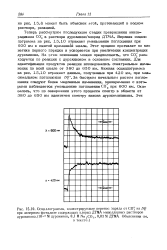
Теперь рассмотрим последующие стадии превращения анион-радикала СОз в растворе дурохинон / хлорид ДТМА. Верхняя осциллограмма на рис. 15.10 отражает уменьшение поглощения при 600 нм в сжатой временной шкале. Этот процесс протекает по кинетике первого порядка и ускоряется при увеличении концентрации дурохинона. На этом основании можно предположить, что СО расходуется по реакции с дурохиноном в основном состоянии. [4]
Образование газа продолжается и на последующей стадии превращения органического вещества - в органогенных илах. [5]
Хотя количественных данных в настоящее время нет, применение скоростной спектрофотометрии свидетельствует о том, что комплексообразо-вание происходит очень быстро и не лимитирует последующих стадий превращения. [6]
В 1953 г. Леви, Тафт и Гаммет [20] показали, что общепринятый механизм гидратации олефинов, изображенный на схеме 6 и состоящий в предравновесном образовании из олефина карбониевого иона с последующей стадией превращения этого иона, определяющей скорость реакции, является непригодным в случае присоединения к изомерным 2-метилбутенам. [7]
Практически это, по-видимому, не так по той причине, что химическое превращение ВВ при горении на начальных стадиях обычно идет с поглощением тепла и тогда не может быть самораспространяющимся. Последующие стадии превращения экзо-термичны; однако, если давление очень мало, то они практически не имеют места, и горение становится невозможным. Если повышать давление, то, начиная с некоторого предела ( зависящего также от внешних условий), процесс становится суммарно экзотермичным и способен к самораспространению. [8]
 |
Петрографический состав исходных углей и твердого остатка ( в %. [9] |
В табл. 2 приведены также результаты определения содержания фенолов и оснований в продуктах растворения углей. Повидимому, фенолы образуются в последующих стадиях превращения тяжелого масла в более легкокипящие продукты. [10]
По технико-экономическим соображениям, как будет показано далее, в аэротенке целесообразно поддерживать возможно более низкую концентрацию кислорода. Таким образом, концентрация растворенного кислорода в иловой смеси около 1 - 2 мг / л обеспечивает нормальное проведение процесса, который контролируется последующими стадиями превращения вещества. [11]
Во всех трех разобранных реакциях наблюдаются некоторые общие черты; наблюдаются, однако, и некоторые различия. Можно считать однозначно доказанным, что на ртутном электроде в кислых растворах механизм этих реакций определяется замедленной стадией присоединения электрона к молекуле или иону реагирующего вещества. Образовавшиеся малоустойчивые промежуточные продукты путем дальнейших реакций превращаются в устойчивые конечные продукты реакции. Эти последующие стадии превращения, невидимому, уже не сказываются на общей кинетике реакции. [12]
Основное направление превращения погребенного органического вещества сапропелевого типа состоит в потере кислорода в виде углекислоты и воды. Само собой разумеется, это не единственное направление реакции, а только преобладающее, потому что одновременно может происходить образование метана, сероводорода и аммиака, что не увеличивает, а снижает запасы водорода и с этой точки зрения является процессом, невыгодным в смысле образования углеводородных смесей. Выделение метана в начальных стадиях превращения вещества сапропелевого типа происходит бактериальным путем и не сопровождается образованием гомологов метана. Эти процессы происходят также и в последующие стадии превращения образовавшейся нефти. Биологическое нроисхождедие метана связано, вероятно, с потерей карбоксильной группы и обязано сопряженным окислительно - восстановительным биохимическим процессам, идущим в точном соответствии с законами термодинамики. Экспериментально показано, что меченый углеродный атом, введенный в карбоксильную группу, выделяется в виде углекислоты или метана, и поэтому, очевидно, бактерии не могут создать более сложные гомологи, нежели метан. Процесс декарбоксилирования может протекать и при низких температурах без участия организмов, но при наличии алюмосиликатных катализаторов, что проверено и опытным путем. При этом возникают новые вещества нейтральной природы как содержащие кислород ( кетоны), так и содержащие только метановые, нафтеновые и ароматические углеводороды. Часть воды образуется при этом за счет ангидридизации кислот, а также при дальнейших превращениях нейтральных кислородных соединений. Термокаталитическое превращение кислот может служить, следовательно, источником углеводородных смесей, характерных для цефти. Жировой материал после гибели организмов прежде всего подвергается ферментативному гидролизу с образованием глицерина и жирных кислот, вероятно, также и окси-кислот, если начальные стадии изменения протекали в доступной кислороду водной среде, что более или менее вероятно при медленном накоплении донных осадков. В дальнейшем оксикислоты теряют воду и превращаются в непредельные кислоты, вступающие в разнообразные реакции по двойным связям и декарбоксилирования. Во всяком случае невозможно предполагать, что декар-боксилирование может сразу дать углеводороды, соответствующие цени углеродных атомов исходной жирной кислоты. [13]
Природа активных центров является дискуссионным вопросом. Различные исследователи считают, что носителями каталитической активности могут быть следующие группировки: 1) гидроксильные группы, способные играть роль бренстедовских кислотных центров, 2) кислородные вакансии или другие дефекты каркаса, которые способствуют появлению льюисовских кислотных центров, 3) многозарядные катионы, создающие градиенты электростатического поля, 4) центры электронного переноса и 5) комбинация различных центров. При изучении активных центров основное внимание обычно уделяется исследованию тех поверхностных группировок, которые могут взаимодействовать с углеводородами с образованием ионов карбония. Значительно меньше ясности в вопросе о том, какую роль играет поверхность цеолитов в последующих стадиях превращения углеводородов в продукты реакции. У большинства исследователей сложилось единое мнение о том, что бренстедовские кислотные центры протонируют молекулы ненасыщенных углеводородов с образованием карбониевых ионов. Эти кислотные центры возникают при термическом разложении ионов аммония или при гидролизе воды под действием многозарядных катионов. Более дискуссионным является вопрос о роли льюисовских кислотных центров. Выяснение этого вопроса, возможно, потребует дальнейших исследований структурных изменений, сопровождающих дегидрокси-лирование и ультрастабилизацию цеолитов. Судя по некоторым работам, льюисовские кислотные центры способны взаимодействовать с ОН-группами и изменять таким образом силу бренстедовских кислоШЫА центров, однако могут ли льюисовские центры непосредственно реагировать с углеводородами и в особенности с парафинами, пока не ясно. В первых работах по катализу на цеолитах высказывалось мнение, что многозарядные катионы также способны взаимодействовать с органическими молекулами с образованием карбониевых ионов. В настоящее время считается, что каталитическая активность цеолитов с многозарядными катионами связана с существованием бренстедовских центров, возникающих при гидролизе воды под действием катионов. Одновременно с гидролизом происходит образование связи между двумя многозарядными катионами через общий атом кислорода, в результате чего стабильность каркаса повышается. При более детальных исследованиях цеолитов с многозарядными катионами было бы интересно выяснить, какую роль в катализе может играть взаимодействие этих катионов с брен-стедовскими или льюисовскими центрами. [14]
Страницы: 1