Введенная вода
Cтраница 3
Рассмотрим начальные участки кинетических кривых. Начальный нестационарный участок может быть связан как с собственной нестационарностью по концентрации активных центров, так и с наличием в системе активного ингибитора, выгорающего по ходу процесса. Таким ингибитором могут быть следы воды. Однако добавки воды не приводят к увеличению индукционного периода, тогда как скорость полимеризации и молекулярный вес полимера уменьшаются пропорционально концентрации введенной воды. Были рассмотрены случаи, когда некий ингибитор может быть внесен в полимеризационную систему с мономером, растворителем и инициатором. Анализ кинетической схемы в этих случаях также приводит к тому, что полученные закономерности не подтверждаются экспериментом. Таким образом, наблюдающиеся индукционные периоды связаны с собственной нестационарностью по концентрации активных центров. [31]
Жидкие фенолальдегидные смолы, получаемые при конденсации примерно эквимолекулярного количества фенола и формальдегида при температурах около 100, в присутствии 0 5 - 1 % щелочей, хотя и содержат около 15 - 20 % воды, только в очень ограниченной степени гидрофильны. Такие смолы представляют собой системы, в которых вода связана со смолой при помощи фенола, щелочи и других сольватизаторов. В соответствии с современными представлениями о коллоидных растворах агрегативная устойчивость этих систем обусловлена образованием мицелл-комплексов из нерастворимого коллоида, стабилизатора и воды. Разбавление этих обычных жидких фе-нолальдегидных смол водой приводит к снижению концентрации стабилизаторов и к разрушению вследствие этого, в зависимости от количества введенной воды и глубины конденсации продукта, того или иного количества мицелл. При этом отслаивается не только введенное количество воды, но и вода разрушенных мицелл. Из обычных фенолальдегидных жидких смол вода отделяется также при хранении их при низких ( 15 - 20) температурах. Нестабильность обычных жидких фенолальдегидных смол к разбавлению и охлаждению усугубляется химической нестойкостью, изменением вязкости и других свойств их в процессе хранения. [32]
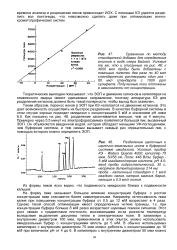 |
Сравнение по методу.| Разделение щелочных и. [33] |
Таким образом, перенос ионов в ЭОП при КЭ налагается на движение катионов. Это дает возможность осуществить их быстрое разделение. В качестве буферной системы в этом случае хорошо подходит имидазол с концентрацией 5 мМ и значением рН ниже 6.0. Как показано на рис. 48, разделение заканчивается меньше, чем за 4 минуты. Примерно через 5.5 минут на электрофореграммах появляется большой пик, вызванный ЭОП. Он объясняется введенной водой, которая обладает меньшим УФ-поглощением, чем буферная система, и тем самым вызывает каждый раз отрицательный пик, с помощью которого можно определить ЭОП. [34]
Хотя фирмы выпускают силикагель возможно более узких фракций, все же товарный силикагель необходимо еще раз поделить на фракции посредством просеивания или седиментации, а затем, если необходимо, промыть разбавленным раствором гидроксида натрия, органическими растворителями, например хлороформом, метанолом, и водой и после этого высушить. Можно проводить дезактивацию, добавляя такие спирты, как пропанол, этиленгликоль, глицерин, но чаще всего дезактивируют силикагель водой. Активность этого адсорбента обычно определяют с помощью азокрасителей [33]; методика определения подробно описана в разд. Соотношение между количеством введенной воды и полученной активностью адсорбента показано в табл. 4.4. В большинстве случаев для хроматографи-рования пригоден адсорбент, содержащий 10 - 12 % воды. Далее мы обсудим способы приготовления сили-кагеля, его разделения на фракции, дезактивации, регенерации, а также пропитки нитратом серебра. [35]
В кружку Эсмарха наливают 1 л воды комнатной температуры и, открыв кран, заполняют ею трубки, вытесняя воздух. Больной ложится на левый бок на край кровати, согнув колени и приведя их к животу для расслабления брюшного пресса. Под ягодицы накладывают клеенку, свободный конец которой опускают в таз на случай, если больной не сможет удержать воду. Наконечник смазывают вазелином, левой рукой разводят ягодицы, а правой легкими вращательными движениями вводят его на глубину 10 - 12 см. Вначале наконечник на 3 - 4 см вводят по направлению к пупку, а затем - параллельно копчику. В течение 5 - 10 минут больной должен удерживать введенную воду, а затем под него подкладыва-ют судно и кишечник освобождается. [36]
После увлажнения катализатора через него вновь пропускали гексан. Затем активность катализатора начала повышаться и через 20 - 25 мин. Следовательно, катализатор при увлажнении заметно не окислился, и понижение содержания ароматических углеводородов вызвано обратимым отравлением его парами введенной воды. [37]
Испытание проводится в два этапа. Электрометрическое титрование проводят определение концентрации воды в испытуемом бензине. На втором этапе испытания, если будет установлено, что концентрация воды в испытуемом бензине летнего вида 0 05 % мае. С помощью медицинского шприца на 1 см3 вводят воду до концентрации 0 05 % в летний бензин и до концентрации 0 15 % - в зимний бензин. Пробу тщательно перемешивают для растворения введенной воды в бензине. [38]
С-22 с нанесенными на него 3Q % нингидрина. Набивку в реакционном сосуде а можно эффективно использовать для пяти отдельных проб. При более низкой температуре реакция обычно не завершается. Хроматографическая колонка б подробно описана ниже при изложении условий для хроматографического разделения. Реакционный сосуд в служит для гидрокрекинга альдегидов до метана и воды. Катализатор сохраняет активность после анализа 50 проб. При работе при пониженных температурах наблюдается значительное размытие хвоста пиков. Осушительная колонка г служит для удаления из газового потока введенной воды и воды, образовавшейся в результате реакции. Набивку следует сменять после пяти опытов. [39]
Последние использовались также в самой методике определения влажности нефтепродуктов. Полученные данные приведены в таблице. Сходимость результатов по определению количества воды в нефтепродуктах, полученных Е разных лабораториях метод м Фишера, не совсем удовлетворительная. Поэтому, к сожалей, трудно по данным результатам установить их достоверность и сопоставить со спектральными данными. Остается путь по определению количества введенной воды. [40]
Последние использовались также в самой методике определения Влажности нефтепродуктов. Предложенный ИК-опектральный метод сопоставлялся о методом Фишера определения влажности нефтепродуктов. Полученные данные приведены в таблице. Фишере, ве совсем удовлетворительная. Поэтому, к сожалению, трудно по данным результатам установить их достоверность и сопоставить со спектральными данными. Остается путь по определению количества введенной воды. [41]
Страницы: 1 2 3