Прямое адсорбционное измерение
Cтраница 1
Прямые адсорбционные измерения показывают также, что адсорбция при пептизации золей происходит избирательно только на определенных участках поверхности. Коллоидные частицы при этом имеют электрический заряд одинакового знака с зарядом длинноцепочечного иона, использованного для пептизации. Поэтому весьма вероятно, что в этом случае адсорбция сводится к образованию на первом, ранее сформировавшемся адсорбционном слое второго слоя с обратной ориентацией за счет вандер-ваальсовых сил взаимодействия между двумя рядами углеводородных цепей. На рис. 101 схематически показано строение адсорбционного слоя при коагуляции и пептизации. [1]
Прямые адсорбционные измерения на платине, являющейся основным электродным материалом в рассматриваемых реакциях, при потенциалах фф0 осложнены описанными выше особенностями окисления поверхности. Тем не менее, весьма полезная информация об адсорбируемости органических веществ в этой области потенциалов может быть получена при комплексном анализе емкостных и кинетических данных. [2]
Применение радиоактивных индикаторов для прямых адсорбционных измерений, естественно, ограничено теми ионами или молекулами, для которых имеются подходящие радиоактивные изотопы. [3]
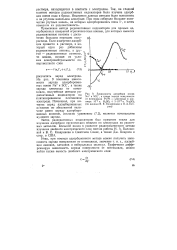 |
Зависимость адсорбции ионов Na и SO -, а также заряда поверхности от потенциала Pt / Pt - электрода в растворе Ю-3 н. H2SO4 3 - Ю-51 н. Na2SO4 ( по данным В. Е. Казаринова и О. А. Пет-рия. [4] |
Применение метода радиоактивных индикаторов для прямых адсорбционных измерений ограничено теми ионами, для которых имеются радиоактивные изотопы с достаточно большим периодом полураспада. [5]
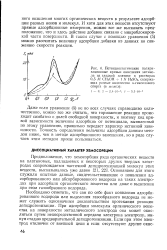 |
Потенциостатические поляризационные кривые окисления метанола на гладкой платине в растворах 0 5 М СНзОН 1 N H2S04, содержащих разные количества н-гексилового спирта ( в моль / л. [6] |
И хотя для этих веществ отсутствуют прямые адсорбционные измерения, можно все же высказать предположение, что и здесь действие добавок связано с микроблокировкой части поверхности. [7]
Полосы поглощения при 1470 см - характерные для спирта, полностью исчезают и появляются интенсивные полосы поглощения смолы, что соответствует прямым адсорбционным измерениям, свидетельствующим о вытеснении спирта с поверхности рутила. [9]
Смещение его, зависящее от полярности связи металл - водород, действительно существует, и в случае платины и родия установлено прямыми адсорбционными измерениями в согласии с выводами из термодинамических соотношений. Однако минимум на кривых дифференциальной емкооти, полученных в [ l2f ], не удовлетворяет указанным выше критериям, и их потенциалы не могут рассматриваться как потенциалы нулевого заряда. [10]
Получаемые в этом случае значения доли гидроксильных групп поверхности, участвующих во взаимодействии с адсорбирующимися молекулами, наиболее близки к получаемым из прямых адсорбционных измерений при том же давлении пара и температуре. [11]
 |
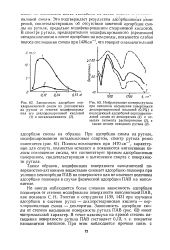
Стационарные поляризационные кривые окисления метанола. [12] |
На рис. 4 пунктиром нанесены зависимости максимальной ( при 6н к 0) скорости адсорбции, выраженной в электрических единицах от потенциала, полученные из независимых прямых адсорбционных измерений в тех же условиях. На рис. 2, 3 и 5 приведены зависимости максимальной скорости адсорбции от потенциала, полученные измерением максимальных ( при т 0) нестационарных токов ионизации водорода. Непосредственное сравнение скорости адсорбции и стационарной скорости электроокисления метанола показывает, что при фг0 65 в скорость адсорбции во всех растворах выше скорости электроокисления и не может являться замедленной стадией процесса электроокисления метанола в стационарных условиях. [13]
Газовая хроматография представляет собой очень удобный метод для измерения количеств реагирующих веществ, действительно адсорбированных на поверхности катализатора во время протекания реакции. Рассмотрим сначала общие соображения. Важно понять, что при определенном давлении данного реагирующего вещества его количество, адсорбированное на катализаторе при равновесии ( определяемое из прямых адсорбционных измерений), может быть несколько меньше или вообще не соответствовать количеству этого реагирующего вещества, фактически адсорбированному во врем: я каталитической реакции при том же самом парциальном давлении. Заполнения поверхности, получаемые во время действительного хода катализа, должны зависеть от механизма реакции и в особенности от той ее стадии, которая определяет скорость процесса. Это в настоящее время совершенно неоспоримо установлено Тамару [532-540], главным образом при изучении реакций синтеза и разложения аммиака на металлических катализаторах. [14]
Страницы: 1