Начальная участка - кинетическая кривая
Cтраница 1
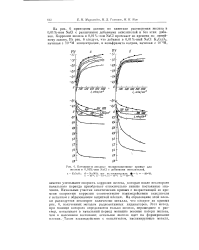 |
Катодные и анодные поляризационные кривые для железа в 0 01 % - ном NaCl с добавками окислителей. [1] |
Начальные участки кинетических кривых с возрастающей во времени скоростью коррозии соответствуют взаимодействию окислителя с металлом с образованием защитной пленки. На образование этой пленки расходуется некоторое количество металла, что следует из кривых рис. 6, полученных методом радиоактивных индикаторов. Этот метод, при помощи которого определяется только железо, перешедшее в раствор, показывает в начальный период меньшие весовые потери железа, чем в пассивном состоянии; остальное железо идет на формирование пленки. [2]
Начальным участкам кинетических кривых, соответствующим стадии образования зародышей графита, отвечают следующие значения энергии активации: для бутана - 300, изобутилена - 280, пропана - 236, пропилена - 224 кДж / моль. [3]
Рассмотрим начальные участки кинетических кривых. Начальный нестационарный участок может быть связан как с собственной нестационарностью по концентрации активных центров, так и с наличием в системе активного ингибитора, выгорающего по ходу процесса. Таким ингибитором могут быть следы воды. Однако добавки воды не приводят к увеличению индукционного периода, тогда как скорость полимеризации и молекулярный вес полимера уменьшаются пропорционально концентрации введенной воды. Были рассмотрены случаи, когда некий ингибитор может быть внесен в полимеризационную систему с мономером, растворителем и инициатором. Анализ кинетической схемы в этих случаях также приводит к тому, что полученные закономерности не подтверждаются экспериментом. Таким образом, наблюдающиеся индукционные периоды связаны с собственной нестационарностью по концентрации активных центров. [4]
При этом начальные участки кинетической кривой накопления полимера аппроксимируются параболой в координатах X - Р ( где X - выход полимера, t - время), типичной для реакций окисления углеводородов, само ускоряющихся за счет радикального распада накапливающихся гидроперекисей. При доступе кислорода воздуха в полимеризующуюся систему интегральный выход полимера ( за 2 5 ч) возрастает почти в 3 раза. Таким образом, существенный вклад в механизм термополимеризации мономеров могут вносить реакции распада образующихся гидроперекисей, вызывающие разветвление цепей. [5]
Таким образом, начальные участки кинетических кривых были аппроксимированы прямой линией. [6]
Применяя способ определения р по начальным участкам кинетических кривых, Я. Л. Забежинский [12] экспериментально исследовал зависимость р от скорости потока, зернения сорбента и некоторых других параметров, полагая, что скорость адсорбции определяется внешней диффузией. Опыты были проведены со слоем сорбента толщиной в одно зерно и единичными зернами. [7]
| |
Величины энергий активации и предэкспоненциальных множителей. [8] |
Поэтому определение констант скоростей обычно ведется или при очень малых концентрациях реагента, или лишь по начальным участкам кинетических кривых, или, наконец, в присутствии каких-либо мономеров, например акрилони-трила, которые реагируют с образующимися свободными радикалами, инициирующими процесс полимеризации, предотвращая тем самым реакции этих радикалов с перекисями. [9]
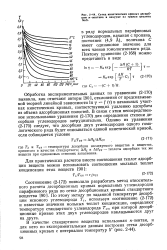 |
Сетка кинетических кривых десорбции н-пентана в вакууме из гранул цеолита NaX. [10] |
Обработка экспериментальных данных по уравнению ( 2 - 170) выявила, как отмечают авторы [90], отклонения от предсказываемой теорией линейной зависимости In 7 f ( т) в начальных участках кинетических кривых, соответствующих удалению адсорбата из объема адсорбционных полостей. В связи с этим непосредственное использование уравнения ( 2 - 170) для определения степени десорбции углеводородов затруднительно. [11]
Методами математической статистики доказаны наиболее вероятные виды уравнений формальной кинетики. По начальным участкам кинетических кривых рассчитаны значения констант скоростей прямых реакций. [12]
Методами математической статистики доказаны наиболее веро-ятные виды уравнений формальной кинетики. По начальным участкам кинетических кривых рассчитаны значения констант скоростей прямых реакций. [13]
Реакцию первого порядка проводят с различными количествами исходного вещества. Пересекутся ли в одной точке на оси абсцисс касательные к начальным участкам кинетических кривых. [14]
На рис. 1 приведены кинетические кривые разложения кумилгидроперекиси в водных растворах бромистых N-цетилпиридиния и N-цетил - З - карбамидопиридиния в слабокислой ( рН 5), нейтральной ( рН 7) и слабоосновной ( рН 9) средах. Расчет скоростей разложения гидроперекиси по начальным участкам кинетических кривых показывает, что в случае бромистого N-цетилпиридиния в слабощелочной среде процесс протекает приблизительно в 400 раз быстрее, чем в слабокислой. В случае бромистого N-цетил - З - карбамидопиридиния скорость распада в щелочной среде примерно в 160 раз превышает скорость разложения ее в слабокислой среде. Вместе с тем распад кумилгидроперекиси в водных растворах бромистого N-цетил - З - карбамидопиридиния протекает значительно быстрее, чем в водных растворах бромистого N-цетилпиридиния. [15]
Страницы: 1 2