Матричный метод хартри
Cтраница 1
Матричный метод Хартри - Фока применим к неэмпирическим расчетам умеренно больших систем. Однако методу Хартри - Фока присущ ряд недостатков. Наиболее серьезным из них является то, что, хотя относительная погрешность вычисленного значения полной энергии невелика, абсолютное значение этой погрешности имеет такой же порядок величины, как самые большие энергии, представляющие интерес для химии. Например, энергия связи для молекулы гидрида лития составляет, - 1, 1 % ее полной энергии, а для молекулы азота - всего 0 2 % ее полной энергии. Кроме того, метод Хартри - Фока приводит к неправильному описанию диссоциации молекул, если частицы, на которые диссоциирует молекула, не обладают замкнутыми электронными оболочками. Даже для простейшей из молекул - молекулы водорода - приближение Хартри - -, Фока не позволяет получить правильного описания диссоциации. Наконец, следует упомянуть, что метод Хартри - Фока часто приводит к плохому описанию спинового распределения вблизи ядер. [1]
Шмидт и Рюденберг [617] выполнили матричным методом Хартри - Фока расчеты для атомов элементов второго и третьего периодов с использованием равномерно сбалансированных базисных наборов гауссовых функций. [2]
Например, при исследовании молекулы азота в рамках матричного метода Хартри - Фока [83] изучена зависимость полной энергии Е, кинетической энергии Tt потенциальной энергии V и орбитальных энергий от качества базисного набора и продемонстрировано, что, хотя полная энергия и орбитальные энергии не очень чувствительны к составу базисного набора, его расширение может приводить к ошибочному поведению кинетической энергии и потенциальной энергии. [4]
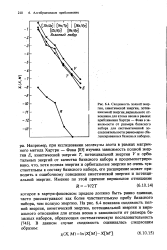
На рис. 7.10 проводится сопоставление сходимости энергии, вычисленной матричным методом Хартри - Фока, и корреляционной энергии для атома бериллия. Очевидно, что при расширении базисного набора корреляционная энергия сходится медленнее, чем энергия, вычисленная матричным методом Хартри - Фока. Использование систематической последовательности расширения базисных наборов, описанной в разд. [5]
В отношении первых двух факторов установлено [474] ( см. также [730]), что некоторые преимущества достигаются при аппроксимации полиномами различного порядка энергий, полученных в рамках модели независимых электронов ( как, например, матричный метод Хартри - Фока), а также корреляционных поправок. [6]
С годами накопился большой материал, позволяющий судить о способности определенных базисных наборов обеспечивать заданную точность в рамках заданной модели. Тот факт, что расчеты матричным методом Хартри - Фока с использованием базисных наборов двухэкспонентного качества приводят к хорошим результатам для длин связей, представляет собой эмпирическое наблюдение, основанное на многих расчетах. Можно было бы утверждать, что такие результаты объяснимы компенсацией между поляризационными эффектами и корреляционными эффектами, но это утверждение невозможно строго обосновать теоретически. Однако из этого вовсе не следует, что подобные эмпирические наблюдения бесполезны. Действительно, выбор базисного набора может рассматриваться как часть концепции теоретической модели химии ( см, разд. Если конкретный метод, использующий определенный базисный набор, был применен к целому кругу проблем и при этом были установлены его точность и применимость к решению этих проблем, то этот метод приобретает определенную предсказательную способность. Подобный подход широко распространен в применениях квантово-механических методик к изучению молекул. [7]
На рис. 7.10 проводится сопоставление сходимости энергии, вычисленной матричным методом Хартри - Фока, и корреляционной энергии для атома бериллия. Очевидно, что при расширении базисного набора корреляционная энергия сходится медленнее, чем энергия, вычисленная матричным методом Хартри - Фока. Использование систематической последовательности расширения базисных наборов, описанной в разд. [8]

В табл. 6.13 иллюстрируется важная роль высших гармоник для молекулярных систем. Здесь представлены результаты расчетов молекулы моноксида углерода с использованием базисных наборов функций s -, р - и d - симметрии, а также базисных наборов функций s -, p -, d - и f - симметрии [748] и показана зависимость энергий матричного метода Хартри - Фока и корреляционных энергий, полученных диаграммным методом теории возмущений, от геометрии ядер. [10]
К иному типу атомных и молекулярных свойств относятся ожидаемые значения. При расчете таких свойств могут приобретать большое значение корреляционные эффекты, которые не играют важной роли при вычислении корреляционной энергии. Так, при определении дипольных моментов оказалось ( см., например, [234]), что в конфигурационном разложении наиболее важную роль играют одночастичные возбуждения, тогда как при вычислении корреляционных энергий наиболее важными оказываются двухчастичные возбуждения. В табл. 3.7 представлены вычисленные в работе [474] дипольные моменты ряда двухатомных гидридов. Результаты, полученные матричным методом Хартри - Фока и в приближении связанных электронных пар, сопоставляются с экспериментальными. [11]
Например, равновесная геометрия ядер молекулы непосредственно определяется электронной энергией. Равновесная геометрия, определенная с использованием ограниченного базисного набора при неполном учете электронной корреляции или вовсе без него, часто приводит к довольно точным значениям длин связей. Такое согласие с экспериментальными данными чаще всего оказывается не случайным. Расширение базисного набора в матричном методе Хартри - Фока обычно приводит к несколько меньшим длинам связи по сравнению с экспериментальными значениями. Учет корреляционных эффектов приводит к возрастанию вычисленных длин связей и в результате к лучшему согласию с экспериментальными значениями. Учет корреляционной энергии имеет важное значение также при определении формы потенциальных кривых и поверхностей. Учет корреляционной энергии может заметно влиять на вычисляемые разности энергий, например барьеры внутреннего вращения или потенциалы ионизации. [12]
Страницы: 1