Данная хроматограмма
Cтраница 2
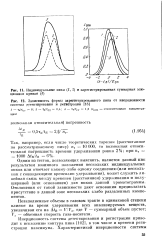 |
Индивидуальные пики ( 1, 2 и зарегистрированная суммарная элю-ционняя кривая ( 3.| Зависимость формы зарегистрированного пика от инерционности системы детектирования и регистрации. [16] |
Одним из тестов, позволяющих выяснить, является данный пик результатом взаимного наложения нескольких индивидуальных пиков или отвечает какому-либо одному соединению ( или нескольким с совпадающими временами удерживания), может служить линейная связь между временем ( расстоянием) удерживания и полушириной ( или основанием) для пиков данной хроматограммы. [17]
Эффективное число тарелок N в отличие от числа п оказывается удобной характеристикой эффективности колонки. Однако эффективное число тарелок неодинаково для всех пиков данной хроматограммы. Вследствие различных коэффициентов диффузии отдельных компонентов оно, так же как и п, зависит от природы компонента. [18]
Первые три пика не разрешены, следующие четыре - хорошо разрешены, а ширина самого последнего пика такая же, как у предыдущих трех разрешенных пиков. Это означает, что последний пик, показанный на данной хроматограмме, может быть в действительности не последним, кроме того, не ясно, имеются ли еще олигомеры, задержавшиеся в колонке и вымываемые позднее в виде очень широких пиков. [19]
Первые три пика не разрешены, следующие четыре - хорошо разрешены, а ширина самого последнего пика такая же, как у предыдущих трех разрешенных пиков. Это означает, что последний пик, показанный на данной хроматограмме, может быть в действительности не последним. [20]
Имеет смысл вкратце обсудить роль теории в хроматографии. Хотя процесс хроматографичеекого разделения чрезвычайно сложен, имеются простые модели и уравнения, которые будут довольно близко аппроксимировать его. Например, если в данной хроматограмме получено недостаточное разрешение, чтобы улучшить его, исследователь должен подобрать новый набор условий. Он может отбирать буквально из бесконечного числа вариаций, и поэтому поиски условий наилучшего разделения ( разрешение в минимальное время) могут длиться довольно долго. Однако уже небольшой запас знаний основ разделения в хроматографии дает возможность логически подойти к подбору условий, а следовательно, облегчить и ускорить его. Даже выбор начальных усло-рий определяется пониманием теории. [21]
Имеет смысл вкратце обсудить роль теории в хроматографии. Хотя процесс хроматографического разделения чрезвычайно сложен, имеются простые модели и уравнения, которые будут довольно близко аппроксимировать его. Например, если в данной хроматограмме получено недостаточное разрешение, чтобы улучшить его, исследователь должен подобрать новый набор условий. Он может отбирать буквально из бесконечного числа вариаций, и поэтому поиски условий наилучшего разделения ( разрешение в минимальное время) могут длиться довольно долго. Однако уже небольшой запас знаний основ разделения в хроматографии дает возможность логически подойти к подбору условий, а следовательно, облегчить и ускорить его. Даже выбор начальных условий определяется пониманием теории. [22]
Времена удерживания, найденные при низких температурах, для всех возможных алканов Сэ указаны на капиллярной хро-матограмме рис. 4; эта хроматограмма отвечает фракции С9 изобутан-бутиленалкилатов. Многие из алканов С9 не входили в состав проб алкилатов, и на хроматограмме изображались пики, полученные с чистыми эталонными соединениями или продуктами метиленирования; это делалось для того, чтобы отметить те места, где должны были бы появиться соответствующие соединения, если бы они находились в пробе. Хотя разделение было хорошим, интерпретация данной хроматограммы сложна, поскольку некоторые изомеры С9 обнаруживают одинаковые времена удерживания. [23]
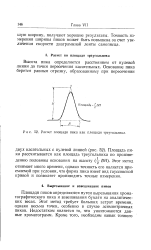 |
Расчет площади пика как площади треугольника. [24] |
Площади пиков определяются путем вырезывания хрома-тографического пика и взвешивания бумаги на аналитических весах. Этот метод требует больших затрат времени, однако весьма точен, особенно в случае асимметричных пиков. Недостатком является то, что уничтожаются данные хроматограмм. [25]
В литературе приводятся величины Rf для многих веществ во многих системах растворителей, однако для аналитиков эти данные могут служить только для ориентировки. Аналитик не всегда способен провести эксперимент в точно таких же условиях, как автор опубликованных данных. Поэтому при идентификации веществ с помощью хроматографии в тонких слоях сорбентов всегда нужно пользоваться растворами свидетелей, подвергшихся аналогичной обработке на данной хроматограмме. [26]
При делении концентрата моноциклических ароматических углеводородов на оксиде алюминия первыми вымываются алкилбензолы с алкильной цепью изопреноидного строения. На рис. 4, а показана хроматограмма фракции 1, являющейся типичной для всех первых фракций, получаемых при делении на оксиде алюминия. На данной хроматограмме наблюдается сравнительно небольшое количество хроматографических пиков. Самый интенсивный пик, обозначенный Аи-27, представлен мети лалкилбензо лом состава С27 с алкильной цепью изопреноидного строения, содержащей 20 углеродных атомов. [27]
Наилучшим методом определения изопреноидных углеводородов является газовая хроматография широкой фракции насыщенных углеводородов, проводимая в режиме линейного программирования температуры с применением высокоэффективных капиллярных колонок. Изопреноидные углеводороды нефтей весьма различны по своему молекулярному весу и содержатся поэтому в различных по температурам выкипания фракциях. С, самый высококипящий - лико-пан - 496 С. На рис. 48 и 49 были приведены хроматограммы фракций, содержащих изопреноиды, и показаны места их элюиро-вания. Однако для того чтобы лучше ориентироваться в порядке элюирования всех 25 алифатических изопреноидов, обнаруженных в нефтях, на рис. 55 представлена унифицированная хромато-грамма, показывающая порядок элюирования изопреноидных углеводородов относительно реперов - алканов нормального строения. Следует обратить внимание, что данная хроматограмма является чисто условной, составленной искусственным путем на основе хроматограмм различных фракций, и не отображает ни относительных концентраций приведенных углеводородов, ни точных значений индексов удерживания. Целью унифицированной хроматограммы является лишь быстрая ориентировка в порядке выхода изопреноидных углеводородов относительно сетки нормальных алканов. Точные же значения концентраций различных изопреноидов рассмотрены ниже. [28]
Метод тонкослойной хроматографии заключается в следующем: на одну сторону небольшой стеклянной пластинки наносят тонкий слой сорбента. На такой слой, так же как на бумагу в бумажной хроматографии, на стартовую линию наносят пробы веществ и их смесей и край пластинки, ниже стартовой линии, погружают в систему растворителей. По мере продвижения жидкости по пластинке происходит разделение смеси веществ. Границу подъема жидкости или линию фронта отмечают, пластинку сушат и проявляют1, подобно бумажной хроматограмме, для обнаружения веществ в виде окрашенных пятен. Отмечают, как указано на рис. 1, положение пятен, отвечающих исследуемым веществам и находящихся между линией старта и линией фронта жидкости. Отношение расстояния от стартовой линии до центра пятна ( отрезок АБ) к расстоянию от стартовой линии до линии фронта ( отрезок АВ) обозначается через константу Rf, характеризующую положение вещества на данной хроматограмме. [29]
По мере продвижения печи вдоль колонки происходит испарение, адсорбция и десорбция компонентов смеси с одновременным ее разделением. Первым начинает выходить из колонки о-ксилол. Его пары, попадая в капилляр, конденсируются и стекают в пробирку. Во время опыта пробирки сменяют после того, как объем жидкости в них составит 0 2 - 0 3 мл. Жидкость в каждой пробирке взвешивают, после чего определяют ее показатель преломления. По полученным данным строят хроматограмму в координатах: масса - показатель преломления. Данные хроматограммы должны указать на полноту разделения смеси и на количество каждого компонента. По этим данным вычисляют состав смеси, или, если ее состав был известен, выход каждого из компонентов. [30]
Страницы: 1 2