Воспроизведение - методика
Cтраница 1
Воспроизведение методики на другом приборе с применением другой партии твердого носителя или неподвижной фазы может привести, при заданных условиях анализа, к получению худших результатов разделения, чем были получены при разработке методики. Иногда разделение может быть улучшено корректировкой анализа, однако формально такая корректировка недопустима. [1]
Нередко возникают трудности при воспроизведении хро-матографических методик, описанных в литературе. Это может быть связано с тем, что носители различаются по плотности. [2]
Фармакопейная статья должна содержать всю информацию, необходимую для однозначного воспроизведения методики, вместе с тем она адресована персоналу, обладающему определенной квалификацией. Поэтому не следует чрезмерно ограничивать те параметры режима, которые не существенны для воспроизведения эксперимента. Например, в данном случае мы не ограничиваем геометрическую характеристику колонки каким-либо одним типоразмером, что сразу создало бы трудности при воспроизведении на приборах различных марок. [3]
В некоторых работах [9] совершенно опускается конкретная характеристика материала носителя, вследствие чего воспроизведение методики становится невозможным. [4]
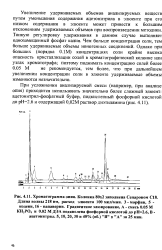 |
Хроматограмма опия. Колонка 80x2 заполнена Сепароном CIS. [5] |
Увеличение удерживаемых объемов анализируемых веществ путем уменьшения содержания ацетонитрила в элюенте при его низком содержании в элюенте может привести к большим отклонениям удерживаемых объемов при воспроизведении методики. Тонкую регулировку удерживания в данном случае выполняет однозамещенный фосфат калия. [6]
Перед проведением статистической обработки результатов важно, чтобы разработчик СО, основываясь на своем профессиональном опыте п квалификации, рассмотрел отчет аттестующей лаборатории и осуществил тщательный анализ полученных данных. При этом следует проверить правильность воспроизведения методики, полноту учета влияния мешающих компонентов, корректность применяемой процедуры контроля. Особое внимание следует обратить на то, чтобы полученные в лаборатории результаты по различным МВИ, были независимыми и укладывались в интервал допустимого разброса результатов, устанавливаемого с учетом приписанных характеристик погрешности аттестованных или стандартизованных МВИ. [7]
Рассмотренная в этой главе совокупность известных данных по влиянию твердого носителя свидетельствует о реальной опасности возникновения систематических ошибок, обусловленных адсорбцией и каталитической активностью используемых твердых носителей, а также реакционной способностью используемых НЖФ. Это необходимо учитывать и на практике при разработке новых и воспроизведении известных газохроматографиче-ских методик. [8]
Метрологические характеристики описываемых методик анализа не приводятся в тексте лишь в тех случаях, когда соответствующие сведения отсутствуют в ори: гинальных публикациях. Критерии оценки ся в этих публикациях даются чрезвычайно редко, поэтому при воспроизведении методик анализа возможно и некоторое несоответствие между приведенными в этой главе данными и экспериментальными значениями сн, установленными по определенному конкретному критерию. [9]
Поэтому чаще всего задача исследователя состоит в установлении эмпирической корреляции между строением исследуемого соединения и получаемым в результате пиролиза спектром низкомолекулярных соединений. В зависимости от природы образца, условий пиролиза и хроматографического разделения продуктов деструкции могут быть реализованы различные спектры. С целью достижения возможности воспроизведения методик в различных лабораториях условия пиролиза и хроматографического разделения должны быть заранее определены и стандартизованы. [10]
Когда у меня возникла мысль о создании многотомного издания, охватывающего важнейшие методы химии углеводов, я не предполагал, что оно будет постоянно пополняться новыми сборниками. В течение долгого времени в наших лабораториях собирались методики, важные для аналитической и препаративной химии углеводов. Некоторые из них были простым воспроизведением прекрасных методик, разработанных другими авторами, другие были модифицированы и усовершенствованы нами в процессе исследований. Первоначально все эти методики были объединены в Книге методов ( Procedure Book), которой широко пользовались наши аспиранты, а позднее сотрудники других лабораторий. От них и поступило предложение составить сборники надежных методов, описанных настолько полно и наглядно, чтобы они были удобны химикам и биохимикам в ординарной лабораторной работе. Эти сборники должны сэкономить специалисту в области химии углеводов время, затрачиваемое на поиски подходящей методики в обширной литературе, и облегчить неспециалисту в области химии углеводов трудную задачу выбора лучшей из нескольких методик. [11]
Исходя из химической природы разделяемых компонентов, хроматографист должен выбрать подходящий состав ПФ и соответствующий но химической природе сорбент. Таким обра: юм, селективность колонки всегда относительна. Она зависит от элюента, типа сорбента, определенное влияние на нее имеют и такие термодинамические факторы, как температура и давление в колонке, изменяющее коэффициенты распределения веществ между ПФ и НФ. Подбор оптимальной селективности является обязательным предметом теоретических и практических изысканий хроматографистов при разработке и воспроизведении методик. [12]
В большинстве случаев хроматографисты пользуются стандартным рядом длин колонок: 25, 15 или 10 см. В последнее время многие фирмы освоили выпуск более коротких колонок ( вплоть до 3 см), заполненных особо мелкозернистыми сорбентами. Однако из теоретических основ метода ясно, что сама по себе длина колонки влияния на качество разделения не оказывает, а ее увеличение способствует увеличению продолжительности разделения. Действительно определяющим фактором является эффективность колонки, и именно ее необходимо указывать, описывая разделение. Это позволяет осознанно подходить к воспроизведению методик разделения и одновременно использовать возможности сокращения продолжительности анализа. Допустим, что, согласно опубликованной методике, разделение выполнялось на колонке длиной 25 см и эффективностью 5000 теоретических тарелок. По современным воззрениям такая колонка не может считаться высококачественной, однако примеров подобного рода в литературе, и даже новейшей, более чем достаточно. В настоящее время для получения указанной эффективности достаточно колонки длиной 10 или даже 5 см. Поэтому имеется реальная возможность, сохранив все остальные параметры опыта постоянными, воспроизвести ранее достигнутое качество разделения на более короткой колонке и за более короткое ( 2 5 - 5 раз) время. [13]
Поэтому при описании результатов хроматографических экспериментов коэффициенты емкости, эффективность, линейная скорость подвижной фазы должны указываться наряду с приведенными выше характе ристиками. Однако из теоретических основ метода ясно, что сама по себе длина колонки влияния на качество разделения не оказывает, а ее увеличение способствует увеличению продолжительности разделения. Действительно определяющим фактором является эффективность колонки, и именно ее необходимо указывать, описывая разделение. Это позволяет осознанно подходить к воспроизведению методик разделения и одновременно использовать возможности сокращения продолжительности анализа. Так, допустим, что согласно опубликованной методике разделение выполнялось на колонке длиной 25 см и эффективностью 5000 теоретических тарелок. По современным воззрениям такая колонка не может считаться высококачественной, однако примеров подобного рода в литературе, и даже новейшей, более чем достаточно. В настоящее время для получения указанной эффективности достаточно колонки длиной 10 см или даже 5 см. Поэтому имеется реальная возможность, сохранив все остальные параметры опыта постоянными, воспроизвести ранее достигнутое качество разделения на более короткой колонке и за более короткое ( в 2 5 - 5 раз) время. Следовательно, выбор длины колонки и эффективности в каждом конкретном случае определяется той селективностью, которой обладает данная система по отношению к разделяемым соединениям, а также требованиями к быстроте разделения. [14]
Если набор эталонных соединений отсутствует, можно попытаться провести идентификацию по табличным значениям величин удерживания. Межлабораторная воспроизводимость абсолютных величин ( tR, k) пока неудовлетворительна. Поэтому множество опубликованных в такой форме данных почти бесполезно при качественном анализе. Тем не менее такой способ идентификации вполне применим по отношению к тем объектам, состав которых хорошо изучен. Лекарственные вещества, а также примеси в них можно отнести именно к такой категории. При воспроизведении методик анализа в этом случае фактически необходимо провести не идентификацию ранее неизвестных веществ, а лишь привязать измеренные величины удерживания к опубликованным величинам удерживания и структурам соединений. [15]
Страницы: 1