Использование - метод - молекулярная динамика
Cтраница 1
Использование метода молекулярной динамики и вариация формы фронта ударной волны позволяют обнаружить ( методом машинного моделирования) возникновение в кристаллическом материале областей с квазижидкой структурой и квазивязким характером поведения, дробление зерен и включений, некристаллографический характер течения материала. [1]
При использовании метода молекулярной динамики в соответствии с заданным силовым полем рассчитывается сила, приложенная к каждой частице, и уравнения движения решаются путем численного интегрирования. После моделирования по методу молекулярной динамики следует перевод системы в реальное-время. Интервал времени между последовательными конфигурациями должен быть достаточно мал, чтобы успевать за самыми быстрыми флуктуациями с частотами порядка 1015 Гц. В результате общее реальное время, которое может быть покрыто при использовании метода молекулярной динамики, в лучшем случае по порядку величины составляет 100 пс. [2]
При использовании метода молекулярной динамики перескоки между различными частями пространства конфигураций исключены, так как каждая конфигурация является механическим следствием предыдущей. В принципе метод Монте-Карло в этом отношении не имеет ограничений. На практике траектория в пространстве конфигураций, которая получается при моделировании методом Монте-Карло, обычно состоит из ряда тесно связанных точек. Так происходит потому, что большие изменения конфигурации, как правило, приводят к неприемлемо большим увеличениям энергии. Требуются специальные методы, которые повышают частоту ввода новых конфигураций в другие части пространства конфигураций. [3]
Такое рассмотрение может быть проведено на основе использования метода молекулярной динамики. В рамках этого метода кристалл рассматривается как система N классических частиц ( атомов), взаимодействующих по определенному закону, который задается потенциалом взаимодействия. [4]
Вычисленные таким образом значения D оказались лишь на 15 - 20 % ниже экспериментально измеренных для аргона при тех же температурах. Расчеты при использовании метода молекулярной динамики ведутся с числом частиц порядка нескольких сотен, максимально около тысячи. Этот метод является наиболее точным для исследования свойств вещества за небольшие промежутки времени и фактически очень близок к прямому эксперименту с реальным веществом, с тем отличием, что мы точно знаем, с какой системой имеем дело, поскольку потенциал взаимодействия задается самим экспериментатором. [5]
В книге помещен обзор научных исследований, выполненных в лаборатории химической термодинамики. Изложены результаты конкретных термодинамических исследований: использование метода молекулярной динамики в теории жидкости и физической кинетики, расчет термодинамических свойств веществ методом молекулярного подобия, масс-опектрометрическое исследование термодинамических свойств веществ, термодинамика дефектообразования в ферритах, определение термодинамических характеристик па-ров некоторых металлов и карбидов, некоторые вопросы термодинамики полимеризации лактанов и др. В ряде обзорных статей отражены результаты исследований, выполненных в лаборатории термохимии им. [6]
По мере увеличения интенсивности УВ закономерности распределения энергии между молекулами и внутри их могут изменяться. Эта гипотеза предложена и обоснована авторами работ [7.15, 7.16], выполненных с использованием методов молекулярной динамики, и подтверждается удовлетворительным совпадением экспериментальных и расчетных результатов [7.16] по моделированию эволюции сильных ударных волн в ВВ. Согласно результатам авторов этих работ скорость передачи энергии молекулам в начале ударного фронта ( в направлении его распространения по кристаллу) становится столь сильной, что энергия не успевает перераспределиться по всем степеням свободы соударяющихся молекул до момента разрыва наиболее быстро возбуждаемой жесткой и прочной связи. До того, как энергия поступательного движения релаксирует и возбудятся наиболее медлительные внутримолекулярные связи, еще внутри фронта У В происходит химическое изменение ВВ, начинающееся с разрыва наиболее прочной и быстро возбуждаемой связи. Следствием этого является образование специфических для сильных УВ ПР во фронте и протекание соответствующих вторичных реакций в потоке за фронтом УВ, отличных от тех, что реализуются при термическом механизме активирования ВВ. [7]
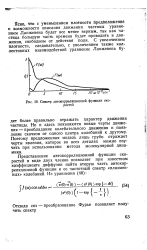 |
Спектр автокорреляционной функции скоростей. [8] |
Но и здесь появляются новые черты движения - преобладание колебательного движения и. Поэтому предложенная модель лишь грубо отражает черты явления, которое во всех деталях можно изучить при использовании метода молекулярной динамики. [9]
Очевидно, что подобная задача может быть решена лишь с помощью ЭВМ. В настоящее время имеются работы ( см., например, обзор [1]), в которых на основе использования метода молекулярной динамики удалось вычислить наблюдаемые величины при числе N элементов макросистемы, достигавшем нескольких сотен. При этом система динамических уравнений, описывающих изменение элементов макросистемы во времени, как правило, представляет собой систему нелинейных дифференциальных уравнений в частных производных. Кроме того, можно отметить, что для нахождения явного вида функций q ( i необходимо было бы также задать значения всех обобщенных координат в начальный момент времени. Это означает, что при описании, например, макросистемы, представляющей собой заключенный в некотором объеме газ, нужно было бы задать координаты и импульсы всех его молекул. [10]
Таким образом, динамическое влияние межцепного взаимодействия на отдельную цепь описывают усредненным коэффициентом трения, и такая динамическая модель пригодна для описания неравновесных свойств сополимера в целом. При этом разумно изучать поведение не реальной макроцепи, а модельной. Расчеты при использовании метода молекулярной динамики ведутся с числом частиц до третьего порядка. Этого достаточно, чтобы система вела себя как макроскопическая. Поэтому изучение молекулярной структуры и решение полученных таким образом уравнений на ЭВМ встречает значительные трудности. [11]
При использовании метода молекулярной динамики в соответствии с заданным силовым полем рассчитывается сила, приложенная к каждой частице, и уравнения движения решаются путем численного интегрирования. После моделирования по методу молекулярной динамики следует перевод системы в реальное-время. Интервал времени между последовательными конфигурациями должен быть достаточно мал, чтобы успевать за самыми быстрыми флуктуациями с частотами порядка 1015 Гц. В результате общее реальное время, которое может быть покрыто при использовании метода молекулярной динамики, в лучшем случае по порядку величины составляет 100 пс. [12]
Страницы: 1