Большая константа - скорость
Cтраница 2
Иначе говоря, процесс последовательного отщепления электрофильной замещающей группы ( или атома галоида) от углеродного атома и обусловленное этим образование смежной двойной связи характеризуется гораздо большей константой скорости, чем возникновение изолированной двойной связи, происходящее по закону случая. [16]
Интенсификация процесса может быть достигнута и при использовании смесей хемосорбентов, что в ряде случаев дает возможность повысить движущую силу процесса [243], а также при использовании хемосорбентов с большей константой скорости [27, 244], например полиаминов. Их преимущество в скорости хемосорбции по сравнению с первичными или вторичными аминами связано также с повышением интенсивности поверхностной конвекции. [17]
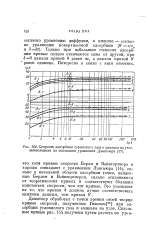 |
Скорость адсорбции сернистого газа и аммиака на угле, вычисленная на основании уравнения Дамкелера ( 27. [18] |
Берля и Вейнгертнера и хорошо совпадают с уравнением Лэнгмюра ( 14), однако в начальной области адсорбции точки, найденные Берлем и Вейнгертнером, сильно отклоняются от теоретических кривых и соответствуют большим константам скорости, чем эти кривые. [19]
Смысл этого действительно заключается в том, что константы равновесия для уравнения (3.5) будут изменяться точно так же, как и константы равновесия для уравнения (3.17), и что большая константа равновесия для реакции (3.5) означает большую константу скорости. Однако нельзя утверждать, что этот вывод является истинным на основании какого-либо закона физической химии. [20]
При совместном решении уравнений ( 176), ( 183) и ( 184) для двух параллельных реакций первого порядка с ki kz, как показано на рис. 16, в условиях хорошего контакта реакция с большей константой скорости дает более высокие степени превращения. Однако при плохом контакте избирательность процесса снижается. Эффективность контакта может быть произвольно определена как отношение веса катализатора, необходимого для достижения определенной степени превращения ( в условиях диффузионного течения), к весу катализатора, необходимого для получения той же самой степени превращения в условиях фактически преобладающего течения. Таким образом, если эффективность контакта невелика, то, чтобы получить ту же самую общую степень превращения, потребуется значительно большее количество катализатора. Наблюдается пропорциональное увеличение степени превращения менее реакционноспо-собного вещества В за счет более реакционноспособного вещества А. [21]
Испочьзун метод цифрового модечирования и допуская, что процесс идет по механизму ЕСЕ, моделировали вольтамперограммы для разчичиьтх значений отношения константы скорости быстрой химической стадии ( k) к скорости развертки ( v) Бычо показано, что вполне реальные значения отношения k / v позволяют получать кривые с предпиком, аналогичным наблюдавшемуся в работе [77] По видимому, для наблюдения таких предпиков необходима большая константа скорости k при ограниченной концентрации нуктгео-фила. [22]
Влияние различных R лучше всего рассмотреть в ряду соединений, у которых группа X сохраняется постоянной. Большая константа скорости kz говорит о неустойчивости молекулы. [23]
Наиболее медленно реагируют с озоном полимеры, содержащие фенильные циклы в главной цепи, в то время как полициклические ( полинафтилены, полиантрацены) или полимеры с гете-роатомами ( поликарбонат) вступают в реакцию значительно легче, В ряду полимеров с насыщенной углеводородной цепью скорость реакции возрастает от полиизобутилена к поливинилцикло-гексану, одновременно наблюдается уменьшение числа разрывов цепи. Самая большая константа скорости у полибутадиена и полиизопрена и у них же наименьшее число разрывов на один акт реакции. Определение констант скорости у соединений, приведенных в табл. 8.1, проводилось аналогично описанным ранее для низкомолекулярных соединений. [24]
Взаимодействие ( А) выбрано в определенной мере как модельное. При такой большой константе скорости даже вблизи точки эквивалентности, т.е. при концентрациях взаимодействующих компонентов меньше 10 М время полупревращения составляет около 0 1 с. Учитывая практически мгновенное установление равновесия в растворе вплоть до точки эквивалентности и обратимость обеих рассматриваемых систем на индикаторных электродах, следует ожидать совпадающие значения потенциалов Р1 и с.э. и классического вида кривые потенциометрического титрования. [25]
 |
Зависимость равновесного потенциала. g ( HB3 системы 1гС11 - 3 - от ионной силы раствора при. [26] |
Это, в частности, наблюдается при перезарядке ионов металлов в системах вида IrClg 3 -, Fe ( CN) g -, Fe ( bpy) 1 2, образованных высшими комплексами окисленной и восстановленной форм. Для приведенных систем характерны большие константы скорости как гомогенных ( см. разд. Поэтому их равновесные потенциалы устойчивы даже в сильно разбавленных растворах с низкими концентрациями окисленной и восстановленной форм ( до 10 6 - 10 - 4 М), что создает благоприятные условия для проверки взаимосвязи между величиной равновесного потенциала редокс-системы и ионной силой раствора [ г в условиях, когда справедлива теория Дебая - Хюккеля. [27]
Иная картина наблюдается, когда в образец 10 не вводят катионы кальция, а образуют декатионированные места. Лантан-декатионированный образец 12 обладает значительно большей константой скорости при низком значении наблюдаемой энергии активации. Таки - м образом, при равной суммарной степени замещения лантан-декатионированный цеолит является более активным, чем лантан-кальциевый. Этот вывод справедлив для всех групп, характеризующихся различным содержанием катионов лантана. [28]
Более активный катализатор В характеризуется большей константой скорости а и меньшим значением температурного коэфициента. Последнее указывает на то, что различие в активности обоих образцов катализатора будет уменьшаться с ростом температуры и катализатор В выгоднее применять для крекинга парафинов при более низких температурах, а катализатор Б - при высоких. При работе с образцом В регулирование процесса следует вести, изменяя объемные скорости, а не температуры; над катализатором Б способ управления крекингом может быть обратным. [29]
Более активный катализатор В характеризуется большей константой скорости а и меньшим значением температурного коэффициента. Последнее указывает на то, что различие в активности обоих образцов катализатора будет уменьшаться с ростом температуры, и катализатор В выгоднее применять для крекинга парафинов при более низких температурах, а катализатор Б - при высоких. При работе с образцом В регулирование процесса следует вести, изменяя объемные скорости, а не температуры; над катализатором Б способ управления крекингом может быть обратным. [30]
Страницы: 1 2 3 4