Коэффициент - активность - зависенок
Cтраница 2
Из всего сказанного о влиянии межионных сил следует, что коэффициенты активности зависят не только от концентрации данного электролита в растворе, но и от присутствия в нем других электролитов. Значения коэффициентов активности понижаются с возрастанием ионной силы раствора, зависящей от концентраций и зарядов всех присутствующих в растворе ионов. [16]
Упругость насыщенного пара является физическим свойством только разделяемых веществ и не зависит от свойств жидкой фазы, тогда как коэффициенты активности зависят от свойств и тех, и других. Поэтому при решении большинства задач разделения следует учитывать различия в упругости насыщенного пара или в температуре кипения разделяемых веществ, а также из различия коэффициентов активности. Соответственно этим различиям определяется и очередность разделения компонентов. [17]
Отмстим, что в общем случае (21.66) и (21.07) лишь неявно определяют криные кип пни л и конденсации, так как коэффициенты активности зависят от оетана. [18]
В основе табл. IX.2, кроме допущения равенства коэффициентов активности ионов калия и хлора, лежит также предположение о том, что коэффициенты активности зависят только от ионной силы. Коэффициенты активности отдельных ионов, приведенные в табл. IX.2, зависят от сделанного предположения. Однако рассчитанные при ее помощи значения / для электролита не зависят от этого допущения. [19]
Фигурные скобки в этом уравнении указывают на то, что приведенные величины являются активностями; их значения легко можно получить, если концентрации ( величины, приводимые в квадратных скобках) настолько малы, что коэффициенты активности зависят только от заряда; они равны единице в случае нейтральных частиц и имеют стандартные значения Дебая - Мильнера для ионов. [20]
Коэффициенты активности зависят от концентрации, а Ка, как указано выше, - от температуры. Поэтому функция концентраций Кс KajKf зависит как от температуры, так и от концентраций. [21]
 |
Коэффициенты активности некоторых электролитов в растворах ( при 298 К.| Коэффициенты активности ионов в водных растворах ( при 298 К. [22] |
Как видно из табл. 8.2, коэффициенты активности меняются в очень широких пределах: в области разбавленных растворов они стремятся к единице, в то время как в области высококонцентрированных растворов они могут достигать единиц, десятков и даже сотен. В области разбавленных растворов ( ниже 0 1 моль / л) коэффициенты активности зависят главным образом от концентрации и заряда ионов, присутствующих в растворе, и мало зависят от природы растворенных веществ. Эта закономерность известна в теории растворов под названием правила ионной силы. Согласно этому правилу, ионы одинаковой зарядности, независимо от их природы, в разбавленных растворах с одинаковой ионной силой имеют равные коэффициенты активности. [23]
Как видно из табл. 16, коэффициенты активности меняются в очень широких пределах: в области разбавленных растворов они стремятся к единице, в то время как в области высококонцентрированных растворов они могут достигать единиц, десятков и даже сотен. В области разбавленных растворов ( ниже 0 1 моль / л) коэффициенты активности зависят главным образом от концентрации и заряда ионов, присутствующих в растворе, и мало зависят от природы растворенных веществ. Эта закономерность известна в теории растворов под названием правила ионной силы. Согласно этому правилу ионы одинаковой зарядности независимо от их природы в разбавленных растворах с одинаковой ионной силой имеют равные коэффициенты активности. [24]
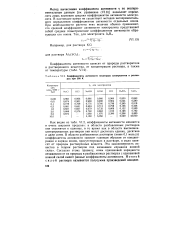 |
Коэффициенты активности некоторых электролитов в растворах при 298 К. [25] |
Как видно из табл. VI.2, коэффициенты активности меняются в очень широких пределах: в области разбавленных растворов они стремятся к единице, в то время как в области высококонцентрированных растворов они могут достигать единиц, десятков и даже сотен. В области разбавленных растворов ( ниже 0 1 моль / л) коэффициенты активности зависят главным образом от концентрации и заряда ионов, присутствующих в растворе, и мало зависят от природы растворенных веществ. Эта закономерность известна в теории растворов под названием правила ионной силы. Согласно этому правилу, ионы одинаковой зарядности независимо от их природы в разбавленных растворах с одинаковой ионной силой имеют равные коэффициенты активности. [26]
Коэффициенты активности, как и парциальные давления паров компонентов, являются функциями состава раствора. Из уравнения ( 1 - 182) следует, что при Т const коэффициенты активности зависят не только от состава раствора, но и от давления, причем эта зависимость проявляется тем сильнее, чем больше изменение объема раствора при смешении и интервал изменения давлений. Поскольку, как указывалось выше, изменение объема при смешении обычно мало, для технических расчетов изменением коэффициентов активности компонентов раствора с изменением давления можно пренебречь и принять, что их зависимость от состава раствора выражается уравнением Гиббса - Дюгема. [27]
Понятие активности иона подменяет понятие его эффективно. Активность иона ч равна произведению сто действительной концентрации С на так называемый коэффициент активности I: а / С. Значения коэффициентов активности зависят от концентрации растворов-ч концентрированных растворах они обычно меньше единицы, а при разбавлении растворов они приближаются к едшшпе Коэффициенты активности можно понимать как меру отклонения от идеальности в поведении вещества в реальном растворе. [28]
В указанных выше случаях определение коэффициентов активности упрощается, так как исследуемые системы можно рассматривать как бесконечно разбавленные растворы, а также потому, что на практике можно постулировать линейные изотермы распределения. Однако если растворенное вещество и растворитель сильно отличаются по полярности, предположение о линейном участке изотермы распределения становится недопустимым даже для области малых концентраций, с которыми встречаются в газо-жидкостной хроматографии; отсюда получается, что удерживаемые объемы, коэффициенты распределения и коэффициенты активности зависят от концентрации растворенного вещества, которая непрерывно уменьшается от входа в колонку к выходу из нее. Эти данные ( табл. 19 и 20) в основном приведены для сравнения; они не отличаются большой точностью из-за отклонения системы от идеальной и связанной с этим отклонением асимметрии кривых проявления. [29]
Страницы: 1 2