Кривая - потенциальная энергия - молекула
Cтраница 2
Для сложных многоатомных молекул потенциальные кривые ( поверхности) неизвестны; однако для качественной характеристики процессов ионизации концепция одномерных потенциальных кривых может быть сохранена. При ионизации различных молекул могут встретиться два случая: 1) кривые потенциальной энергии молекулы и иона им еют минимумы при одинаковых равновесных расстояниях, 2) минимум кривой потенциальной энергии иона находится при несколько большем или меньшем расстоянии, чем в молекуле. [16]
Приближение Борна - Оппенгеймера придает кривой потенциальной энергии молекулы вполне определенный смысл. Если меняется конфигурация ядер, меняется и энергия молекулы, а зависимость энергии от их положения и есть кривая потенциальной энергии молекулы. Для двухатомной молекулы эта кривая представляет собой график зависимости энергии от длины связи, а для многоатомной молекулы вместо кривой получается сложная поверхность. Если нарушить равновесную конфигурацию молекулы, она вернется в положение равновесия ( или по крайней мере будет колебаться около него), следовательно, возрастание энергии при изменении равновесной конфигурации соответствует приобретению молекулой потенциальной энергии. Если бы приближение Борна - Оппенгеймера было неудовлетворительным ( так должно быть в случае легких или быстро движущихся ядер), понятие поверхности потенциальной энергии, равно как и длины связи и углов между связями, потеряло бы смысл. На самом деле это приближение нарушается, но лишь слегка, что вызывает небольшие спектроскопические эффекты. [17]
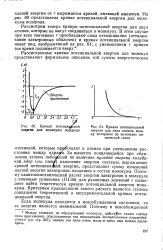 |
Кривая потенциальной энергии для молекулы водорода.| Кривая потенциальной энергии для двух атомов, между которыми не возникает химической связи. [18] |
За вычетом появляющейся при сближении атомов небольшой по величине нулевой энергии колебаний ядер ( см. ниж) изменение энергии системы, выражаемое кривой потенциальной энергии, представляет сумму изменений полной энергии электронов, входящих в состав молекулы. Поэтому квантовомеханический расчет энергии электронов в молекуле с помощью уравнения (111.36) для различных значений г позволяет найти теоретически кривую потенциальной энергии молекулы. Критерием правильности такого расчета является степень совпадения теоретической и экспериментальной кривых потенциальной энергии. [19]
Даже наилучший расчет по методу Хартри - Фока не дает точной электронной энергии атома или молекулы. Что такое корреляционная энергия, почему ее так называют. Если кривая потенциальной энергии молекулы вычислена по методу Хартри - Фока, то как равновесные длины связей и частоты колебаний будут отличаться от их действительных значений. Более или менее важными становятся эффекты, связанные с пренебрежением электронной корреляцией, при удлинении связи между двумя атомами. [20]
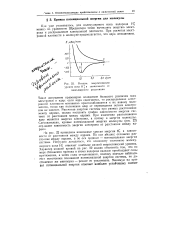 |
Низшие энергетические. [21] |
Такое допущение правомерно вследствие большого различия масс электронов и ядер: если ядра сдвигаются, то распределение электронной плотности мгновенно приспосабливается к их новому положению, тогда как положение ядер от перемещения легких электронов не зависит. При изменении расположения яде ]) меняется энергия электрона, а потому и энергия молекулы. Следовательно, кривые потенциальной энергии молекулы ( рис. 22) отражают зависимость энергии электрона от расстояния между яд-рама. [22]
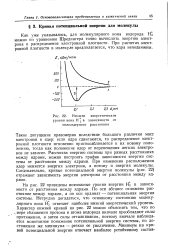 |
Низшие энергетические. [23] |
Такое допущение правомерно вследствие большого различия масс электронов и ядер: если ядра сдвигаются, то распределение электронной плотности мгновенно приспосабливается к их новому положению, тогда как положение ядер от перемещения легких электронов не зависит. При изменении расположения ядер меняется энергия электрона, а потому и энергия молекулы. Следовательно, кривые потенциальной энергии молекулы ( рис. 22) отражают зависимость энергии электрона от расстояния между ядрами. [24]
Вопрос о возможности корреляции структуры и масс-спектра на сегодняшний день не ясен. В неорганической масс-спектрометрии отсутствует классификация масс-спектров и закономерностей их изменения. Что касается теории возникновения масс-спектра простейших Неорганических молекул, то она принципиально ясна и определяется принципом Франка-Кон - дона, а также формой и взаимным положением кривых потенциальной энергии молекулы и молекулярного иона. [25]
Основная химическая информация, которую можно получить при изучении колебательных спектров, сводится к идентификации веществ путем сравнения их колебательных спектров, рассматриваемых как своеобразные отпечатки пальцев. Можно получить и количественную информацию о жесткости связей относительно растяжения и изгиба; соответствующие силовые постоянные являются важными характеристиками химических связей. Ангармоничность колебаний показывает, насколько истинный потенциал отличается от идеального параболического. Колебательная и вращательная структуры электронных спектров дают информацию того же типа о возбужденных электронных состояниях молекул. Эти сведения позволяют получить полную картину кривых потенциальной энергии молекул в различных электронных состояниях. [26]
Франка - Кондона и другими правилами отбора. На колебательную структуру накладывается дополнительная структура, обусловленная возбуждением вращательного движения, при этом наблюдаются главным образом Р - и R-ветви, а в некоторых случаях и Q-ветви. Поскольку в основном и возбужденных электронных состояниях момент инерции отличается, ветви могут образовывать кант ( см. рис. В. Колебательные линии сгущаются в сторону большей энергии, так как колебательные уровни сходятся по направлению к пределу диссоциации. В некоторых случаях колебательная и вращательная структуры исчезают и вновь появляются, прежде чем достигается предел диссоциации, - это проявление предиссоциации. Из спектра можно определить силовые постоянные, энергию диссоциации и ангармоничность электронных состояний молекулы, а также параметры кривых потенциальной энергии молекулы. [27]
Страницы: 1 2