Нулевая линия - хроматограмма
Cтраница 2
Когда газообразный компонент с вязкостью, отличной от вязкости газа-носителя, вводится в колонку, сопротивление колонки, а следовательно, и скорость потока по всей колонке изменяется. Это вызывает изменение разности давлений между точками аи б ( см. рис. 13) и смещение нулевой линии хроматограммы. [16]
В детекторах этого типа газ, ныходящий из колонки, смешивается с Н; и сгорает в кислороде ( иди воздухе), в результате чего происходит ионизация газа в пламени. Предел детектирования определяется в мкг / с, так как чувствительность этих детекторов пропорциональна массовой скорости потока; АЛЯ того чтобы привести это значение к единицам массы ( мкг), его следует умножить на ширину пика ( с) па нулевой линии хроматограммы. [17]
Таким образом хроматография-это физический метод разделения смеси веществ, основанный на различной сорбционной способности компонентов смеси и осуществляемый путем многократного их распределения между подвижной ( газ-носитель) и неподвижной ( насадка колонки) фазами. В результате анализа получается х р о м а тог р а м м а - график зависимости величины сигнала детектора от времени или объема газа-носителя. Нулевая линия хроматограммы соответствует выходу из колонки чистого газа-носителя; пик - выходу одного из компонентов смеси. [18]
Однако по сравнению с усилителями, применяемыми в обычных электрометрических измерениях, к устройствам, используемым в хроматографии, предъявляются дополнительные требования: способность к долговременной работе, высокая линейность и очень малый дрейф фонового сигнала. В особых случаях длительность хроматографического опыта с использованием капиллярных колонок может достигать 14 час. Если полагать, что за время опыта смещение нулевой линии хроматограммы в результате дрейфа усилителя не должно превышать 1 % шкалы регистратора, то при обычном диапазоне шкалы 0 - 1 мв скорость дрейфа не должна превышать 10 мкв в сутки. Это требует высокой стабильности работы источников питания и всех элементов схемы применяемых усилителей. [19]
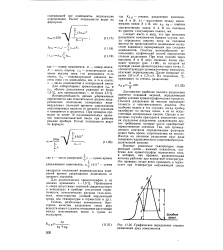 |
Графическое определение степени разделения двух компонентов. [20] |
Следует иметь в виду, что при неполном разделении компонентов бывают случаи, когда определить ширину пика на половине высоты не представляется возможным вследствие взаимного перекрывания зон соседних компонентов. При этом способе через вершины А и В двух смежных пиков проводят прямую ( рис. 11.26), из которой на нулевую линию хроматограммы через точку С, соответствующую промежуточному минимуму, опускают перпендикуляр. [21]
 |
Схема термокон-дуктометрического детектора. [22] |
Детектор представляет собой неуравновешенный мост постоянного тока, плечи которого изготовлены из тонких платиновых проволок, имеющих одинаковое электрическое сопротивление. Плечи R1 и R4 помещены в рабочую камеру, через которую протекает газ, выходящий из разделительной колонки, а плечи R2 и R3 - в сравнительную камеру, через которую пропускается чистый газ-носитель. Все плечи моста нагреваются от включенного в диагональ аЪ источника постоянного тока Б с регулировочным реостатом Др. Реостат R5, расположенный в вершине моста, служит для корректировки нулевой линии хроматограммы, записываемой вторичным прибором. [23]
 |
Параметры пика хроматограммы. [24] |
На рис. 19 изображена характерная форма пика, полученного с детектором дифференциального типа. Нулевая линия записывается регистратором, когда из колонки выходит газ-носитель. Площадь пика - площадь треугольника, равная произведению его полуоснования HI на высоту ЕВ. Отрезок h от нулевой линии хроматограммы до вершины пика называют высотой пика. Измеряют ее масштабной линейкой. Отрезок HI называют шириной пика. Ее замеряют на расстоянии, равном половине высоты пика, с помощью измерительной лупы. [25]
Гидрирование олефшшоносульфонатов проводят ври атмосферном давлении в растворе 50 % - ного этанола при 50 С в присутствии катализатора - Pd ( 5 %) на угле. Окончание реакции гидрирования контролируют по исчезновению полосы поглощения при 965 см 1 на ИК-спектре. На хроматограмме ( жидкая фаза 3 % SE-30) получают четко разделенные пики каждого из гомологов указанных производных. Недостатками метода являются необходимость создания условий, обеспечивающих минимальное термическое разложение сульфонилхлоридов, а также введения поправочных коэффициентов для расчета количественного состава по данным хромато-траммы. Присутствие в исходной пробе дисульфонатов приводит к искажению нулевой линии хроматограммы и снижает точность расчета. [26]
Измерение полного ионного тока может быть выполнено различными способами. Самый простой состоит в использовании коллектора полного ионного тока. Последний представляет собой диафрагму, располагаемую между ионным источником и масс-анализатором, которая виньетирует периферийную область пока еще не диспергированного пучка ионов. Эта диафрагмированная часть ионного пучка служит мерой полного ионного тока. Измеряемый сигнал после усиления его электрометрическим усилителем регистрируется компенсационным самописцем в виде хроматограммы. Если ионный источник, как обычно, работает в режиме энергии электронов 70 эВ, то при этом ионизируется также и газ-носитель, что вносит существенный вклад в полный ионный ток. Для того чтобы произвести правильную запись хроматограммы, эту часть полного ионного тока нужно электрически скомпенсировать. В этих условиях даже незначительные, практически почти неустранимые изменения потока газа-носителя или компенсирующего напряжения вызывают сильную нестабильность нулевой линии хроматограммы и, следовательно, потерю чувствительности детектирования сигнала. От этого недостатка можно в существенной мере избавиться, понизив энергию электронов ионного источника до 20 эВ и выбрав гелий в качестве газа-носителя. [27]
Страницы: 1 2