Квантовомеханическая метода
Cтраница 2
В качестве iF0 в методе согласования могут быть использованы также значения силовых постоянных, найденные по квантовомеханическим методам. Точность их определения по этим методам, как правило, еще недостаточна для того, чтобы их можно было непосредственно включить в итерационную процедуру уточнения по методу наименьших квадратов. [16]
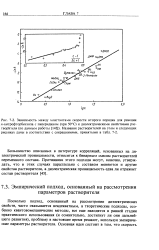
Поскольку подход, основанный на рассмотрении диэлектрических свойств, часто оказывается неадекватным, а теоретические подходы, особенно квантовомеханические методы, все еще находятся в ранней стадии практического использования ( и сомнительно, достигнут ли они дальнейшего развития), проблему в настоящее время решают, используя эмпирические параметры растворителя. [18]
Одна из опасностей такого подхода состоит в необходимости точного определения небольшой разницы между двумя большими величинами, вычисленными приближенными квантовомеханическими методами. Это относится к теплотам образования реагентов и теплоте образования переходного состояния. Поэтому очевидно, что небольшие несистематические ошибки в определении полных теплот образования будут радикально влиять на предсказание величин теплот активации. [19]
В настоящий момент единственный способ обойти трудности - это отказаться от всех попыток абсолютного расчета энергий возбуждения и осторожно применить простые квантовомеханические методы к очень приближенным волновым функциям для грубой оценки небольших изменений в энергии. Это могут быть энергетические изменения в результате введения заместителей или искажения формы молекулы, или изменения в характеристических энергиях перехода в ряду алкенов или полиенов. Применяемые в этом случае волновые функции ( как функции возбужденных состояний, так и волновые функции основного состояния) почти всегда строятся из минимального числа атомных орбиталей основного состояния, причем нет универсального метода - ни более простого, ни сложного. [20]
В настоящий момент единственный способ обойти трудности - это отказаться от всех попыток абсолютного расчета энергий возбуждения и осторожно применить простые квантовомеханические методы к очень приближенным волновым функциям для грубой оценки небольших изменений в энергии. Это могут быть энергетические изменения в результате введения заместителей или искажения формы молекулы, или изменения в характеристических энергиях перехода в ряду алкенов или полиенов. Применяемые в этом случае волновые функции ( как функции возбужденных состояний, так и волновые функции основного состояния) почти всегда строятся из минимального числа атомных орбиталей основного состояния, причем нет универсального метода - ни более простого, ни сложного. Хотя они точно определяют соответствующую им симметрию состояния, однако получаемые результаты оказываются совершенно бесполезными для вычисления, например, силы осциллятора или дштольных моментов. [21]
В настоящее время ( особенно в химии алкенов) накопившиеся спектральные данные по энергии возбуждения можно интерпретировать и систематизировать лишь квантовомеханическими методами, однако точные квантовомеханические расчеты возбужденных состояний даже для простейших алкеновых молекул, вероятно, еще многие годы будут представлять значительные трудности. [22]
В настоящее время ( особенно в химии алкенов) накопившиеся спектральные данные по энергии возбуждения можно интерпретировать и систематизировать лишь квантовомеханическими методами, однако точные квантовомехаиические расчеты возбужденных состояний даже для простейших алкеновых молекул, вероятно, еще многие годы будут представлять значительные трудности. [23]
Прежде всего следует отметить, что даже в том случае, если мы располагаем вычислительными машинами и можем рассчитать достаточно точно межмолекулярные силы непосредственно квантовомеханическими методами, создание приближенной теории межмолекулярных сил представляется весьма целесообразным. Причина этого состоит в том, что полный квантовомехани-ческий расчет при всей своей сложности и трудоемкости позволяет получить лишь численную информацию применительно к конкретной системе частиц, и в этом состоит полезный выход такого расчета. Любая другая система рассматривается как совершенно новая задача, и все расчеты нужно проводить с самого начала. Приближенные методы дают очень грубые численные результаты, тем не менее они достаточно просты и позволяют понять физический смысл явления, скрытый при точных расчетах, производимых на основании тщательно разработанной теории. [24]
В некоторых случаях этот метод, оказавший неоценимые услуги в биологии, является единственным, дающим однозначное суждение о течении химического процесса. Квантовомеханические методы, например метод молекулярных орбиталей ( МО ЛКАО - молекулярные орбитали - линейная комбинация атомных орбиталей), позволяют рассчитывать молекулярные диаграммы органических соединений, включающие такие параметры, как порядок связей, индексы свободных валентностей, эффективные заряды на атомах, и оценивать способность молекул к химическим реакциям. [25]
Ни один из этих методов не может быть применен для всего широкого диапазона, в котором изменяется реакционная способность и селективность в вышеупомянутых типах химических реакций. Квантовомеханические методы расчета поверхностей потенциальной энергии реакций все еще слишком грубы для учета тонких различий в скоростях или направлениях ориентации в зависимости от небольших изменений в заместителе. В любом случае получение всех точек, необходимых для точного определения изменения энергии при изменениях углов и длин связей в трехмерном пространстве, является очень трудоемким и дорогостоящим занятием. Тем не менее тонкие детали механизмов реакций, касающиеся синхронности и природы элементарных стадий, не могут быть получены никаким другим путем. Например, недавние расчеты энергетических поверхностей для циклофрагментации циклобутана с образованием этилена [31] и для геометрической изомеризации циклопропана [32] показали, что промежуточным бирадикальным частицам ( например, тетраметилен и триметилен) не соответствуют минимумы на поверхности потенциальных энергий этих реакций. [26]
Очевидно, что в настоящее время одной из центральных задач теории кристаллов является разработка достаточно точных методов квантовомеханических расчетов энергии межатомной связи и межатомных расстояний в молекулах и кристаллах применительно к многоэлектронным атомам. Существующие квантовомеханические методы приближенные и основаны на сведении задачи к одноэлектронному приближению или на методе аппроксимации в случае многоэлектронного приближения. Оба эти метода, как правило, приводят к сравнительно точным результатам, но таят в себе большие опасности, связанные с необходимостью непрерывного и строжайшего учета оценок допускаемых погрешностей. При расчетах энергий и длин межатомных связей искомые величины определяются как малые разности больших величин и поэтому допускаемые неточности без надлежащего контроля приводят к существенным погрешностям. Большой интерес представляют абсолютные квантовомеханические расчеты молекул и кристаллов с учетом всех электронов. В этом отношении заслуживают внимания, например, работы Пройса, статья которого помещена в настоящем сборнике. [27]
Поскольку в таком переходе промежуточные стадии не всегда доступны экспериментальному исследованию, теоретические предсказания могут оказаться единственным источником информации. Между тем разные квантовомеханические методы расчета приводят к сильно различающимся результатам, касающимся как скорости приближения свойств кластера к свойствам массивного кристалла, так и геометрии стабильных промежуточных конфигураций атомов. [28]
В настоящее время теория взаимного притяжения молекул развивается на основе методов статистической физики. Начинают использоваться и квантовомеханические методы. [29]
Кинетика той же реакции была рассмотрена Вилером, Топлеем и Эйрингом [2] сточки зрения теории абсолютных скоростей химических реакций. Прежде всего, используя квантовомеханические методы, описанные в предыдущей главе, они вычислили энергию активации. Рассчитанное значение оказалось на 10 ккал выше экспериментального; как уже говорилось, такие расчеты действительно приводят только к очень ориентировочным значениям. [30]
Страницы: 1 2 3 4