Определение - потенциальная функция
Cтраница 2
Переход к плоской задаче позволяет применить теорию аналитических функций комплексного переменного для определения потенциальной функции в замкнутой области по значению нормальной производной на ее границах. [16]
Наиболее важная, но и наиболее трудная задача в молекулярной теории адсорбции - определение потенциальной функции Ф взаимодействия молекул с поверхностью твердого тела. При однозначном определении этой функции Ф из опытных адсорбционных данных и при строгих ее кванто-вомеханических расчетах встречаются серьезные трудности. Вместе с тем приближенные, основанные на достижениях полуэмпирической теории межмолекулярных взаимодействий, расчеты потенциальных функций Ф для взаимодействия молекул разного строения с однородной поверхностью многих твердых тел, использующие физико-химические свойства адсор-бата и адсорбента, приводят к значениям термодинамических характеристик адсорбции, находящимся в удовлетворительном согласии с опытом. Особенно хорошие результаты получены при взаимодействии различных молекул с поверхностью неполярных твердых тел - неспецифических адсорбентов, когда основными силами притяжения являются дисперсионные. В случае сложных молекул, состоящих из нескольких сортов силовых центров, например молекул углеводородов, для повышения точности приближенных расчетов Ф в настоящем этапе, по-видимому, целесообразно производить уточнение параметров потенциальных функций ф взаимодействия силовых центров молекулы с поверхностью, используя опытные адсорбционные данные для небольшого числа молекул, состоящих из тех же силовых центров. Эти уточненные потенциальные функции ф далее могут быть использованы для предсказания энергий и термодинамических характеристик адсорбции других молекул, состоящих из этих же силовых центров на том же адсорбенте. [17]
Наиболее важная, но и наиболее трудная задача в молекулярной теории адсорбции - определение потенциальной функции Ф взаимодействия молекул с поверхностью твердого тела. При однозначном определении этой функции Ф из опытных адсорбционных данных и при строгих ее кванто-вомеханических расчетах встречаются серьезные трудности. Вместе с тем приближенные, основанные на достижениях полуэмпирической теории межмолекулярных взаимодействий, расчеты потенциальных функций Ф для взаимодействия молекул разного строения с однородной поверхностью многих твердых тел, использующие физико-химические свойства адсор-бата и адсорбента, приводят к значениям термодинамических характеристик адсорбции, находящимся в удовлетворительном согласии с опытом. Особенно хорошие результаты получены при взаимодействии различных молекул с поверхностью неполярных твердых тел - неспецифических адсорбентов, когда основными силами притяжения являются дисперсионные. В случае сложных молекул, состоящих из нескольких сортов силовых центров, например молекул углеводородов, для повышения точности приближенных расчетов Ф в настоящем этапе, по-видимому, целесообразно производить уточнение параметров потенциальных функций ф взаимодействия силовых центров молекулы с поверхностью, используя опытные адсорбционные данные для небольшого числа молекул, состоящих из тех же силовых центров. Эти уточненные потенциальные функции ф далее могут быть использованы для предсказания энергий и термодинамических характеристик адсорбции других молекул, состоящих из этих же силовых центров на том же адсорбенте. [18]
IX - XI полученные для нулевого заполнения значения термодинамических характеристик адсорбции использованы для полуэмпирического определения потенциальных функций межмолекулярного взаимодействия и для сопоставления с результатами моле-кулярно-статистических расчетов. [19]
Задача определения напряженного состояния шара при заданных поверхностных перемещениях или при заданных поверхностных силах сводится также к обычным задачам определения потенциальной функции по заданным условиям на границе. [20]
Таким образом, при решении задач моделирования полей характерным является то, что параметры системы распределены в пространстве непрерывно и определение исследуемой потенциальной функции ф требует задания положения рассматриваемой точки внутри системы. Поэтому в подобных задачах пространственные переменные х, у и z, а также время t являются непрерывно меняющимися независимыми переменными, а уравнения, описывающие систему - дифференциальными уравнениями в частных производных. Конечно-разностная аппроксимация частных производных по существу представляет собой замену системы с распределенными параметрами набором дискретных элементов, количество которых определяется шагом математической сетки. [21]
Кривые 1 - 4 близки друг к другу и только кривая 5 значительно отличается. Однако кривая 5 наименее точна, так как определению потенциальной функции из опытных данных по внутримолекулярным взаимодействиям мешает неопределенность формы и неточность величины потенциального барьера для внутреннего вращения звеньев СН2 вокруг связи С-С. Все более надежные потенциальные функции вандерваальсового взаимодействия двух атомов углерода близки друг к другу. [22]
Полученные так атом-атомные потенциальные функции далее могут быть использованы для определения потенциальной функции Ф и на ее основе расчета термодинамических характеристик адсорбции при нулевом заполнении поверхности для других соединений рассматриваемого класса на том же адсорбенте с погрешностью, близкой к погрешности соответствующих экспериментальных значений. Полученное хорошее согласие между значениями Ki, рассчитанными при использовании атом-атомного приближения, и экспериментальными значениями Ki для адсорбции на ГТС всех рассмотренных соединений, молекулярная структура которых хорошо известна, позволило поставить и решить обратную задачу молекулярной теории адсорбции - на основании термодинамических характеристик адсорбции определить или уточнить структурные параметры молекулы. [23]
В заключение вкратце остановимся на тех упрощениях, которые получаются при определении потенциальной функции в предположении, что явление движения одноразмерное. [24]
К сожалению, допущение об идеальности раствора не всегда приемлемо. В большинстве случаев приходится применять более общий подход, заключающийся в определении потенциальной функции взаимодействия между разнородными молекулами. Обычно используются потенциальные параметры е, а и иногда ци, как это описано в разделе II. [25]
Все существующие в настоящее время методы определения структуры молекул имеют определенные ограничения. Поэтому поиск новых методов определения структуры сложных молекул, и в особенности поиск новых методов определения потенциальных функций внутреннего вращения сложных молекул, остается актуальной задачей. [26]
Для потенциала ф взаимодействия силовых центров молекулы и твердого тела обычно принимается та же модель, что и для взаимодействия двух простых молекул в газе. При современном состоянии теории межмолекулярных сил использование приближения ( VIII2), по-видимому, дает пока единственную возможность какого-либо прогресса при определении потенциальных функций Ф взаимодействия сложных молекул с учетом зависимости Ф от их ориентации. [27]
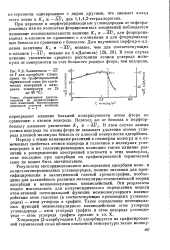 |
Зависимость - ДС7 от Г для адсорбции хлоро-прена на графитированной термической саже ( из статических измерений в интервале температур от 25 до 80 С. [28] |
Результаты экспериментального исследования адсорбции моно - и полигалогенпроизводных углеводородов, помимо значения для идентифицирования в аналитической газовой хроматографии, необходимы для дальнейшего развития молекулярно-статистическои теории адсорбции и межмолекулярных взаимодействий. Такое определение потенциальных функций межмолекулярного взаимодействия атом углерода углеводорода - атом углерода графита и атом водорода углеводорода - атом углерода графита сделано в гл. [29]
Однако, в отличие от прямой задачи, решение обратной задачи представляет весьма сложную проблему, которую до настоящего времени нельзя считать окончательно разрешенной. Удовлетворить этому условию нелегко. Значительные успехи в определении потенциальных функций весьма сложных органических молекул обусловлены использованием концепции переносимости силовых постоянных, применение которой может быть оправдано широким распространением свойства структурной аддитивности, благодаря чему потенциальные функций большого числа различных молекул могут быть построены с помощью ограниченного набора силовых постоянных, для определения которых можно использовать данные о спектрах всех этих молекул. Для неорганических соединений структурная аддитивность не характерна, в связи с чем нет оснований заранее предполагать возможности перенесения силовых постоянных, характеризующих одинаковые структурные элементы в различных молекулах. Определение потенциальных функций сравнительно простых многоатомных молекул с числом атомов от трех до пяти-шести существенно облегчается при использовании в дополнение к данным о частотах колебаний информации о постоянных центрифугального искажения, кориоли-сова взаимодействия и среднеквадратичных амплитудах колебаний. [30]
Страницы: 1 2 3