Пересечение - потенциальная кривая
Cтраница 4
Для двухатомных молекул, где имеется лишь один геометрический параметр - межъядерное расстояние R, в общем случае система уравнений ( 3) будет несовместна, откуда следует утверждение о том, что потенциальные кривые двухатомных молекул не пересекаются. Поэтому более точная формулировка правила непересечения такова: потенциальные кривые двух состояний одного и того же типа симметрии, как правило, не пересекаются, тогда как кривые состояний различных типов симметрии пересекаться могут. Наличие пересечения потенциальных кривых соответствует ситуации, изображенной на рис. 9.1.1 а, однако, как правило, они должны вести себя так, как показано на рис. 9.1.16. Точки R0, где кривые I и II сближаются, называют обычно точками псевдопересечения ( англ. Правило непересечения было установлено Л. Д. Ландау и К. [46]
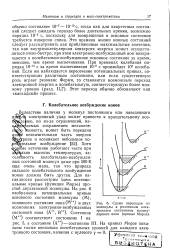 |
Схема переходов от молекулы к различным электронным состояниям молеку. [47] |
Эти времена жизни ионных состояний следует рассматривать как приближенные максимальные значения, так как внутримолекулярная конверсия энергии может осуществляться очень быстро; при этом электронно-возбужденный ион возвращается в колебательно-возбужденное основное состояние за время, меньшее, чем время высвечивания. Если же наблюдается пересечение потенциальных кривых, соответствующих различным состояниям, или если существенную роль играет резонанс Ферми, то становится возможным переход электронной энергии в колебательную, соответствующую более низкому уровню ( разд. Этот переход обычно обозначают как каскадный процесс. [48]
Вычисление потенциального барьера водородных связей с доведением его до конкретных числовых значений остается до сих пор неразрешенной задачей первоочередного значения. Для вычисления барьера в водородных связях функция Морзе крайне ненадежна из-за неточности на больших расстояниях от минимума. Высота барьера, определяемая точкой пересечения потенциальных кривых двух состояний АН - j - В и А НВ, очень чувствительна даже к небольшим изменениям значений параметров, взятых из опыта, особенно межъядерных расстояний. Между тем эти параметры приходится находить из опытных данных, принимая разные упрощающие допущения. [49]
Вычисление потенциального барьера водородных связей с доведением его до конкретных числовых значений остается до сих пор неразрешенной задачей первоочередного значения. Для вычисления барьера в водородных связях функция Морзе крайне ненадежна из-за неточности на больших расстояниях от минимума. Высота барьера, определяемая точкой пересечения потенциальных кривых двух состояний АН В и А НВ, очень чувствительная даже к небольшим изменениям значений параметров, взятых из опыта, особенно межъядерных расстояний. Между тем эти параметры приходится находить из опыт, ных данных, принимая разные упрощающие допущения. [50]
Кривая 1 описывает физическую ( ван-дер-ваальсову) адсорбцию, для нее характерен незначительный минимум на относительно большом расстоянии от поверхности металла. Кривая 2 с глубоким минимумом в точке А указывает на гораздо более прочную связь хемосорбированного атома водорода с поверхностью. В этом случае сила связи водорода с поверхностью металла превышает энергию диссоциации D и адсорбированные молекулы водорода расщепляются, образуя соединения Me-H. Пересечение потенциальных кривых показывает, что физически адсорбированная молекула, которая приобрела анергию, эквивалентную энергии в точке В, может перейти на кривую 2 и хемосорбироваться в виде атома водорода. [51]
 |
Кривые потенциальной энергии основного S0 и возбужденного S, состояний двухатомной молекулы АВ. [52] |
По принципу Франка - Кондона наиболее вероятным будет такой переход, при котором не изменяется ни положение ядер, ни импульс. Наиболее вероятное межъядерное расстояние для молекулы с нулевой колебательной энергией соответствует средней точке CD или RS. Для более высоких колебательных уровней области наибольшей вероятности расположены вблизи точек пересечения потенциальных кривых горизонтальными линиями. Будут наблюдаться и другие, как более длинноволновые, так и более коротковолновые переходы, имеющие меньшую вероятность. С уменьшением межъядерного расстояния потенциальная кривая становится круче, поэтому интенсивность в спектре поглощения спадает в направлении коротких волн более полого, чем с длинноволновой стороны от максимума. [53]
 |
Кривые потенциальной энергии основного So и возбужденного Si состояний двухатомной молекулы АВ. [54] |
Изменение потенциальной энергии электронно - возбужденного состояния в зависимости от расстояния между ядрами атомов представлено кривой Si. Энергия связи в возбужденном состоянии уменьшается по сравнению с основным состоянием, поэтому положение равновесия в возбужденном состоянии отвечает большему межъядерному расстоянию, нежели в основном состоянии. Наиболее вероятное межъядерное расстояние для молекулы на нулевом колебательном подуровне соответствует средней точке CD или RS. Для более высоких колебательных подуровней наиболее вероятные межъядерные расстояния соответствуют точкам пересечения потенциальных кривых с горизонтальными линиями, соответствующими энергии колебательных подуровней. [55]
 |
Кривые потенциальной энергии основного 50 и возбужденного Si состояний двухатомной молекулы АВ. [56] |
По принципу Франка - Кондона наиболее вероятным будет такой переход, при котором не изменяется ни положение ядер, ни импульс. Наиболее вероятное межъядерное расстояние для молекулы е нулевой колебательной энергией соответствует средней точке CD или RS. Для более высоких колебательных уровней области наибольшей вероятности расположены вблизи точек пересечения потенциальных кривых горизонтальными линиями. Будут наблюдаться и другие, как более длинноволновые, так и более коротковолновые переходы, имеющие меньшую вероятность. С уменьшением межъядерного расстояния потенциальная кривая становится круче, поэтому интенсивность в спектре поглощения спадает в направлении коротких волн более полого, чем с длинноволновой стороны от максимума. [57]
Страницы: 1 2 3 4