Бром-катион
Cтраница 1
Присоединение бром-катиона по месту двойной связи с образованием бромгидрииа постоянно нарушает равновесие процесса гидролиза, сдвигая его вправо, и продукты реакции содержат преимущественно гало-генированный спирт. Благоприятное влияние в этом отношении оказывает повышение концентрации хлора или брома. В среде же растворителя, который повышает растворимость галогена, а тем самым и его концентрацию, например в растворе соответствующего алкилендигалогенида или иного растворителя, в котором растворяется образующийся галогенгидрин, подавляется гидролиз галогена, поэтому такой прием но рекомендуется. [1]
Невидимому, бром-катион появляется и в метанольном растворе. [2]
Если же бром-катион взаимодействует с одним из периферийных углеродных атомов ( положения 1 или 4), то образуется карбокатион аллилъного типа, в котором положительный заряд, как в аллильном радикале неспаренность электрона ( см. разд. [3]
Таким образом, концентрация бром-катиона чрезвычайно мала, но его реакционная способность исключительно велика. [4]
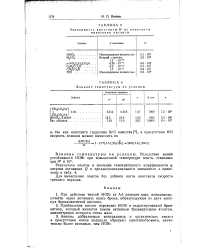 |
Влияние температуры на реакцию. [5] |
Прибавление кислот переводит НОВг в гидратированный бром-катион, который является самым активным бронирующим агентом, концентрация которого очень мала. [6]
Из наших опытов следует, что бром-катион имеет значение и для реакции присоединения по двойной связи. [7]
Этот результат можно объяснить в предположении, что бром-катион на первой стадии реакции образует связанный с обоими атомами углерода симметричный я-комплекс - бромониевый ион. [8]
Гидроксил-катион может присоединяться к двойной связи совершенно аналогично бром-катиону. При этом промежуточно образующийся ион карбония стабилизируется взаимодействием с соседними группами, которое, наконец, переходит в нормальную гомеополярную связь в эпоксиде. Суммарный процесс по необходимости представляет собой цис-присоединение. [9]
При 300 бромирующим агентом ( как и в других реакциях галоиди-ровяния ароматических и гетероциклических соединений) является бром-катион, и бромирование представляет собой реакцию электрофильного замещения; при высоких же температурах ( порядка 500) бромирующим агентом является уже атомарный бром и реакция протекает как радикальное замещение. [10]
Можно исключить свободное вращение вокруг простой углерод-углеродной связи промежуточного карбониевого иона, если допустить, что сначала присоединение бром-катиона Вг происходит сразу к обоим углеродным атомам двойной связи. Последующая атака бром-анионом должна, следовательно, происходить с противоположной стороны двойной связи. [11]
Рассмотрим механизм этой реакции, предположив для простоты, что поляризация во всех образующихся в процессе превращения комплексах доходит до конца и что первоначально молекулу углеводорода атакует бром-катион. [12]
По этой причине молекулярный бром ( слабая кислота Льюиса) реагирует с толуолом в уксусной кислоте более чем в 600 раз быстрее, чем бензол, тогда как бром-катион из гипобромита ( сильная кислота Льюиса) мало различает реакционноспособ-ный толуол и менее реакционноспособный бензол, ре агируя с толуолом лишь в 36 раз быстрее, чем с бензолом ( см. табл. 7.9, а также величины для реакций дифенила, стр. [13]
Реакция толуола с бромом в присутствии бромида железа ( Ш) протекает по совершенно другому механизму. Здесь реагентом не является ни молекулярный, ни атомарный бром. Эта реакция является обратимой, и стационарная концентрация бром-катиона в системе никогда не бывает высокой ( как, впрочем, и в случае с атомарным бромом), но она вполне достаточна, чтобы эффективно вести основную реакцию. [14]
Наоборот, реагент с сильным и остролокализованным электронным дефицитом сравнительно мало нуждается в помощи ароматического соединения, чтобы успешно присоединяться к нему. Напротив, он сам в значительной мере обладает необходимой энергией. По этой причине молекулярный бром ( слабая кислота Льюиса) реагирует с толуолом более чем в 600 раз быстрее, чем с бензолом, в то время как бром-катион из бромноватистой кислоты ( сильная кислота Льюиса) лишь слабо различает активный толуол и менее активный бензол и реагирует с толуолом только в 36 раз быстрее, чем с бензолом ( см. табл. 69, стр. Теперь понятно, что избирательная способность электрофиль-ного реагента распространяется не только на различные типы субстрата, но и в равной мере на различные реакционноспо-собные положения внутри конкретного ароматического соединения. Поэтому в случае богатого энергией ( обладающего большим электронным дефицитом) электрофнльного реагента следует говорить о возможностях стабилизации в переходном состоянии или в ff - комплексе при орто -, мета - или пара-замещениях в меньшей мере, чем в случае бедного энергией электро-фильного реагента, который в значительной степени нуждается в поддержке полярными эффектами заместителей в ароматическом соединении. Другими словами, в случае бедных энергией ( слабокислотных) электрофильных реагентов следует всегда ожидать очень значительного орго / пара-замещения при полном отсутствии или малой доле жега-продукта или, наоборот, практически исключительного замещения в мета-поло-женке в зависимости от заместителей, находящихся в ароматическом соединении. Напротив, в случае богатых энергией ( сильнокислотных) электрофильных реагентов даже при наличии заместителей, способствующих своими полярными эффектами стабилизации переходного состояния для орто / парс-замещеиия, можно всегда ожидать значительного количества жега-произ-водного, и, наоборот, при наличии лгега-ориеитирующих заместителей - также орто - и пара-продукты. [15]
Страницы: 1 2