Построение - поверхность - потенциальная энергия
Cтраница 2
Методы CNDO и INDO плохо воспроизводят теплоты атомиза-ции и орбитальные энергии, в связи с этим они неприменимы для изучения относительной устойчивости различных молекулярных структур, а следовательно, для построения поверхностей потенциальной энергии и изучения механизмов реакций. [16]
На рис. 27.11 а показаны результаты расчета изменений энергии при атаке атомом водорода молекулы водорода под различными углами; в каждом случае связи релак-сируют до оптимальной длины, Как предполагалось при построении поверхности потенциальной энергии на рис. 27.8, при линейной атаке энергия активации будет наименьшей. [17]
Все эти данные необходимы для полного теоретического расчета скорости реакции. Построение поверхности потенциальной энергии является первым этапом такого расчета. [18]
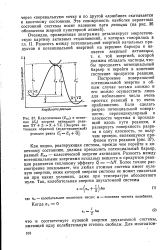 |
Классическая (. кл и истинная (. о энергии активации реакции XY Z - X YZ. [ Энергия активации обратной ( эндотермической реакции равна. j. 0 QJ. [19] |
Очевидно, приведенная диаграмма детализирует энергетическую картину активных столкновений, о которых говорилось в гл. Построение поверхностей потенциальной энергии в общем случае весьма сложно и его можно осуществить лишь для небольшого числа очень простых реакций и то очень приближенно. [20]
 |
Механизм фотохимической реакции превращения диоксетана в формальдегид. [21] |
Экспериментально обнаружено, что реакция превращения диоксетана в формальдегид приводит к возникновению хемифос-форесценции. Построение поверхностей потенциальной энергии данной реакции в основном и возбужденных синглетном и трип-летном состояниях позволяет объяснить природу этого эффекта. На рис. 124 приведены кривые потенциальной энергии реакции. Кривая потенциальной энергии реакции основного состояния пересекается с соответствующей кривой триплетного возбужденного состояния. Вследствие этого реакция может идти двумя независимыми каналами; адиабатическим с образованием двух молекул формальдегида в основном состоянии и неадиабатическим с возникновением одной молекулы формальдегида в возбужденном триплетном состоянии. Сильная интеркомбинационная конверсия вследствие большого спин-орбитального взаимодействия приводит к тому, что реакция идет почти полностью по неадиабатическому каналу. Этим объясняется большой выход молекул формальдегида в триплетном состоянии с последующим высвечиванием путем фосфоресценции. [22]
 |
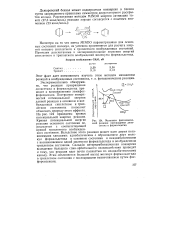
Система трех частиц, лежащих на прямой линии. [23] |
Для построения поверхности потенциальной энергии с указанными выше свойствами угол между осями координат, вдоль которых отложены расстояния между атомами X и Y и между Y и Z, должен определяться из условия, чтобы кинетическая энергия материальной точки, выраженная через соответствующие прямоугольные координаты, представляла собой сумму двух чисто квадратичных членов, без перекрестных членов. [24]
Поэтому для построения поверхности потенциальной энергии необходимо учесть взаимное влияние всех атомов. Для построения поверхности потенциальной энергии даже для реакции молекула - атом не существует точных методов расчета. Применяется полуэмпирический метод расчета, принцип которого сводится к следующему. [25]
Электронные состояния реакционного комплекса рассчитываются для межмолекулярных расстояний, изменяющихся от бесконечности до конечного расстояния связывания. Это ведет к построению поверхностей потенциальных энергий для основных и возбужденных состояний реакций. Можно легко идентифицировать переходные состояния и промежуточные продукты; можно также определить относительную энергетику различных направлений реакции. [26]
В настоящей главе на примерах моделирования различных физико-химических процессов рассмотрены основные этапы исследования методом классических траекторий. Обсуждаются также полуэмпирические и эмпирические методы построения поверхностей потенциальной энергии и численные методы, используемые при динамических расчетах. [27]
 |
Длины аксиальных н экваториальных связей ( ни, рассчитанных в переходных состояниях ( X реакций М СН4 и F CH3F. [28] |
ПЭ в области минимумов, что позволяет с удовлетворительной точностью рассматривать силовые постоянные и частоты нормальных колебаний молекул. В связи с этим метод MINDO / З широко применяют для построения поверхностей потенциальной энергии и исследования механизмов реакции. В то же время метод обладает некоторыми недостатками, которые сужают область его применения. [29]
В схеме метода MINDO / 3 предусмотрена оптимизация геометрии молекулы с помощью градиентного спуска в энергетический минимум. В связи с этим метод MINDO / 3 является наилучшим полуэмпирическим методом для построения поверхностей потенциальной энергии и исследования механизмов реакции. [30]
Страницы: 1 2 3