Представление - молекула
Cтраница 2
Конечно, при строгом рассмотрении, описание спектра 3-метилтио-фена в приближении жесткого волчка даже для переходов с малым / неприменимо вследствие наличия внутреннего вращения. Однако небольшая величина расщепления наблюдаемой тонкой структуры спектральных линий свидетельствует о том, что высота потенциального барьера заторможенного внутреннего вращения довольно большая. Поэтому представление молекулы в виде жесткого волчка как первое приближение, облегчающее дальнейшую идентификацию переходов, является целесообразным. [16]
Один из выводов предыдущего раздела настолько важен, что его следует обсудить более подробно. Предположим, что мы нашли такой набор, в котором атомные орбитали атомов перекрываются попарно, так, что каждая орбиталь значительно перекрывается с орбиталью какого-то другого атома, и слабо - со всеми остальными орбиталями в молекуле. В этом случае представление молекулы соответствующим набором двухцентро-вых локализованных орбиталей должно оказаться удовлетворительным во всем, что касается коллективных свойств молекулы. Хотя этот принцип, который можно назвать принципом локализованных связей, очевиден, ему не всегда придают должное значение. Очень часто теоретическое описание молекулы оказывается чрезмерно усложненным в результате неудачного выбора орбиталей базисного набора. [17]
Система, описанная в работе [6], является дальнейшим развитием предыдущей в том плане, что учитывается пространственное строение молекул. Как и ранее, синтез ведется от конца к началу ( от продуктов реакции к исходным веществам) по заранее определенному набору химических реакций. В работе [8] для представления молекулы в качестве параметров используются тип атома и топо-тогическая структура связей между атомами в молекуле. При том акцент сделан на типы атомов углерода в молекуле в соответ-твии с природой связи углерода с другими элементами. В работе 11 ] для характеристики молекулы используются три параметра: : естоположение атома в молекуле, ковалентные связи между томами и свободные электроны в каждом атоме молекулы. [18]
Образ нашего мышления в органической химии меняется в соответствии с усложнением используемых нами модельных представлений. Представление молекул в виде твердых шарообразных атомов, связанных друг с другом стержнями, было и остается важным в работе химика-органика. Однако, чтобы понять механизмы многих реакций, необходимо более усложненное представление молекул, которое дается теорией электронных пар Льюиса, очень успешно развитой английскими химиками сэром Робертом Робинсоном и сэром Кристофером Ингольдом. Для обозначения смещений электронов в ходе химической реакции были использованы изогнутые стрелки, что привело к значительно лучшему пониманию тех факторов, которые контролируют химические реакции. [19]
Однако этот аргумент несостоятелен. Энергия спектрального перехода равна ( в представлении МО) разности энергий двух молекулярных орбит рассматриваемого соединения. Это свойство зависит от энергий отдельных МО. Представление молекулы с помощью МО совершенно не соответствует классическому изображению молекул с локализованными связями; последнее является искусственным представлением, означающим возможность преобразования МО в набор почти невзаимодействующих эквивалентных орбит, локализованных между парами атомов. Возможность такого преобразования не связана непосредственно с энергиями отдельных МО. Поэтому изменения поглощения света не могут дать никаких сведений о фиксации связей. Это становится очевидным при рассмотрении бутадиена с помощью метода теории возмущений, описанного в гл. Невозмущенная система содержит две локализованные этиленовые п-связи, каждая из которых включает дважды заполненную п - МО. [20]
В основе его лежит представление о молекулярном движении в жидкости, которое отличается от устаревшего ван-дер-ваалъсовского понимания жидкостей как систем, родственных газам. Согласно этим представлениям молекулы жидкости совершают колебания около положения равновесия, как это происходит в твердом теле. В последние годы эти представления были подтверждены данными рентгеновских исследований. Однако, в отличие от твердого тела, в жидкости происходит более частый обмен местами, который обусловливает текучесть. Обмен местами требует перехода через энергетический барьер величиною и. Это время определяет коэффициент самодиффузии D, который равен D ( V6) ( бУт) ( Л) ( б2 / То) ехр ( - u / kT), где 6 - расстояние между рассматриваемыми двумя местами. Это уравнение довольно хорошо описывает температурный ход вязкости в случае простых - не по-лимеризованных - веществ. [21]
Не превышает его и отношение силовых постоянных и энергий водородных связей ( 1 5), образованных двумя атомами водорода молекулы воды. На самом деле оно может быть значительно меньше. Поэтому предлагавшееся ранее [388] представление молекул воды в жидкой фазе в виде двух ОН-осцилля-торов, колеблющихся практически независимо один от другого, следует признать несостоятельным. [22]
Описание не содержит никаких сведений относительно стереохимии соединения. Система ЛФВ позволяет быстро находить данное соединение в большом массиве данных, но не очень хорошо приспособлена для компьютерного планирования синтеза. В программе SYNCHEM-2 развита собственная система линейного представления молекул - SLING. Для манипуляций со структурами программа SYNCHEM использует представление молекул в виде матриц смежности, адаптированное таким образом, чтобы сделать превращение в систему ЛФВ более простым. Кроме матриц смежности, КПОС использует для описания структуры соединения так называемые таблицы связности. Наиболее часто используют форму записи, предложенную Морганом [293] и улучшенную У инке [182, 184] и Моро [294], для включения стереохимических данных. [23]
Рассмотрим теперь математическое представление реактантов, учитывающее явление геометрической изомерии. Отметим сразу, что современные формулы строения химических веществ непригодны для проведения расчетов на ЭВМ химических реакций, так как их нельзя непосредственно ввести в оперативную память ЭВМ или записать на внешние носители информации. Далее, для этой цели нецелесообразно использовать и векторное представление молекул, которое строилось на основе их брутто-формул. Следовательно, требуются дальнейшие обобщения, связанные с представлением молекул в виде матриц определенной размерности, равной числу содержащихся в молекуле атомов. При формировании элементов этой матрицы, называемой jB - матрицей, учитывается, что каждый атом состоит из атомного остова, составленного из ядра атома и внутренних электронов и имеющего некоторый формальный заряд, и электронов валентной оболочки. Последние менее сильно связаны с атомным остовом и участвуют в образовании химических связей. [24]
В случае систем, проявляющих свойство хиральности, возможны две ситуации. Если все конформеры, представленные на МКГ, хиральны, последний может быть разбит на два эквивалентных подграфа-дерева разрезом ребер, соответствующих взаимопревращениям пар энантиомеров. Если система содержит как хиральные, так и ахиральные конформеры, подобное разбиение проводится разрезом таких же ребер и удвоением вершин, соответствующих ахи-ральным конформерам. Такие разбиения и представляют собой учет стереохимической конфигурации при представлении молекул в рамках теории графов. Введя верхнюю границу FMax по высоте барьеров, можно разделить МКГ на подграфы, соответствующие геометрическим изомерам. [25]
Автоматизированный синтез маршрутов протекания химических реакций более всего развит применительно к органической химии. Первый основан на том, что маршруты строятся на основе известных реакций для отдельных стадий, а второй - на генерации промежуточных элементов, образующих дерево маршрутов получения целевых продуктов реакции. Различные ветви дерева являются альтернативными вариантами, которые будут отвергаться или приниматься пользователями. Известны практические реализации этих подходов в виде комплексов программ [6-9], отличающиеся способом представления молекул вещества и стратегией синтеза альтернативных маршрутов реакций. [26]
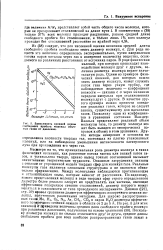 |
Зависимость средней длины свободного пробега молекул обычных газов от давления. [27] |
Несмотря на то, что принципиальная роль диаметра молекул в таких физических явлениях, как рассеяние потока частиц или вязкий поток газов, и установлена твердо, однако вывод точных соотношений вызывает значительные теоретические трудности. Основные затруднения связаны с отказом от упрощенной модели твердых сфер для молекул газа. Реальные молекулы газа являются сложными структурами и не являются обязательно сферическими. Между молекулами наблюдаются притягивающие н отталкивающие силы, которые зависят от расстояния. По-видимому, вместо представления молекул в виде твердых сфер строго определенного диаметра более реально следует их представлять как частицы, имеющие эффективное поперечное сечение столкновений, диаметр которого может меняться в зависимости от типа проводимого эксперимента. Na, Ar, CH4, COa и паров Н2О диаметры эффективного поперечного сечения лежали в области от 2 до б А. [28]
Данный метод не получил еще достаточно широкого распространения в применении к теории жидкого состояния, поскольку расчеты равновесных свойств жидкости требуют минимизации свободной энергии, а не потенциальной, для чего необходим очень большой объем вычислений. В этих работах для описания взаимодействий молекул жидкости чаще всего применялись потенциалы твердых сфер и реже - потенциалы типа 6 - 12 и 6-ехр. При этом даже такие жидкости, равновесная структура которых существенно зависит от ориентации молекул, обычно рассматриваются как одноатомные. Так, в работе [47] методом Монте-Карло были рассчитаны термодинамические функции жидкой воды с потенциалами 6-ехр. Понятно, что представление молекул воды в виде сфер не может быть адекватным, в особенности, если речь идет не о термодинамических функциях, а о равновесной конфигурации жидкости. [29]
Если молекулы находятся достаточно далеко друг от друга, то их взаимная потенциальная энергия равна нулю, а полная энергия W этой консервативной системы равна их кинетической энергии Wt. К моменту максимального сближения молекул ( rri) вся их кинетическая энергия оказывается полностью израсходованной на совершение работы против сил отталкивания [ И / ( г1) 0 ], а их взаимная потенциальная энергия Wn ( r) W. Поэтому в первом приближении можно считать, что г зависит только от химической природы газа и представляет собой не что иное, как эффективный диаметр d молекул. Из сказанного ясно, что возможность представления молекул газа в виде твердых шариков диаметра d связана с очень быстрым увеличением сил взаимного отгалкивания молекул реального газа при уменьшении расстояния между ними. [30]
Страницы: 1 2 3