Природа - алкильный заместитель
Cтраница 1
Природа алкильного заместителя, по-видимому, оказывает незначительное влияние на скорость перегруппировки. Заместители в остатке сахара также, по-видимому, не оказывают значительного влияния на скорость перегруппировки. [1]
Природа алкильного заместителя R ( метил или н-пропил) не оказывает существенного влияния на К в случае изомеризации сульфоксидов и, по-видимому, во всех остальных случаях. В системах, где R - н-пропильная группа, величина константы равновесия, по существу, характеризует относительную способность групп X - СН2 и X стабилизировать систему путем взаимодействия с двойной углерод-углеродной связью. Этот факт нельзя также объяснить перекрыванием р-орбиталей двойной связи с d - орбиталями атома серы, поскольку такое перекрывание должно в случае каждой пары делать более устойчивой винильную, а не аллильную форму, тогда как на деле наблюдается обратное. [2]
В табл. 14 показано влияние изменения природы алкильных заместителей. Большая величина р / 50 -говорит о большей эффективности ингибитора. Совершенно ясно, что высказанное выше предсказание не оправдалось. [3]
Скорость превращения ангы-комплекса в сын-комплекс зависит от природы алкильного заместителя. [4]
Из данных таблицы следует: I) природа алкильных заместителей в молекуле диалкилдитиофосфатов почти не сказывается на величине К. При сравнении К, диалкилдитиофосфатов цинка и ди-4 - трет-октилфенил ( 2 2-диметиленамин) ди-тиофосфата бария ( 3aL2) в окисляющихся алкилароматических углеводородах видно, что 3oL2 примерно на порядок эффективнее комплексов цинка. [5]
Поскольку длительность выдержки при галоидировании фенолов зависит от строения и природы алкильных заместителей в ядре, то этим методом нельзя пользоваться для идентификации фенолов неизвестного строения. [6]
Анализ продуктов окисления с помощью ГЖХ [403] или реакционной ГЖХ [404] позволяет точно установить природу алкильных заместителей в порфинном макроцикле. [7]
В ряду кетонов с точно определенной степенью и положением замещения ( например, ряды Вудворда) природа алкильного заместителя практически не влияет на интенсивность перехода в УФ-спектре. [8]
Мостиковые углеводороды по масс-спектральным свойствам очень близки конденсированным соединениям. Во всех случаях удается определить природу алкильных заместителей, но не всегда их расположение. [9]
Присадки имеют сравнительно низкий мол. Используют пять типов присадок [3], различающихся лишь природой алкильного заместителя в метакри-ловом эфире - н-бутил -, н-гексил -, децилоктил -, лаурил - и стеарил-метакрилаты. [10]
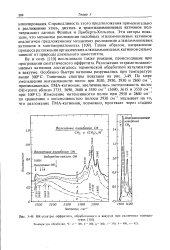 |
ИК-спектры оффретита, обработанного в вакууме при различных температурах. Температура обработки, С. а - 100, б - 200, & - 300, г - 350, д - 400, с-425. [11] |
Справедливость этого предположения применительно к разложению этил -, диэтил - и триэтиламмониевых катионов подтверждают данные Фрипья и Ламберта-Хельзена. Таким образом, направление процесса разложения органических алкиламмониевых катионов сильно зависит от природы алкильного заместителя. [12]
Многие бактерии и грибы продуцируют пиррольные тримеры, в молекулах которых существуют одновременно два упомянутых типа межъядерных связей. В основе их структуры лежит скелет продигиозена 6.67, Его производные называются продигиозанами и отличаются друг от друга природой алкильных заместителей в положениях С2 и СЗ. Атом С6 обычно несет метоксильную группу. Типичный представитель продигиозин имеет химическую структуру 6.68. Это вещество и его аналоги окрашены в красный цвет и относятся к физиологически активным пигментам. Они обладают противомикробной, цитотокеической и фунгипидной активностями, но высоко токсичны и не пригодны для использования в качестве терапевтических препаратов. Кроме того, продигиозины подавляют иммунный ответ у млекопитающих. В этом, видимо, состоит их естественная функция, помогающая бактериям и грибам противостоять защитным реакциям инфицируемого организма. [13]
В настоящем случае содержание А2 - изомсра в первоначально полученной смеси после дегидратации может быть оценено в 42 э от общего количества непредельных сложных эфп-ров, тогда как в равновесной смеси содержится около 67 % сопряженного соединения 3, Таким образом, количество сопряженного изомера в смеси может быть в значительной степени увеличено путем изомеризации непредельных эфиров в присутствии оснований, взятых в качестве катализаторов. Для других непредельных сложных эфиров состав смеси после дегидратации3 7, а также отношение обоих изомеров в равновесной смеси3 8 могут изменяться в зависимости от положения и природы алкильных заместителей в цепи, причем достижение равновесия не всегда может оказаться желательным. Для соответствующих соединений с успехом можно воспользоваться методикой, приведенной ниже, причем существенным является тщательное предохранение от попадания влаги. [14]
Разнообразно замещенные эфиры реагируют в присутствии хлористого алюминия. Принимая во внимание, что с однозамещенными эфи-рами входящая ацильная группа направляется в пара-положение к эфирной связи, ориентация входящей группы при ацилировании замещенных эфиров зависит от природы алкильного заместителя. [15]
Страницы: 1 2