Введение - фенильная группа
Cтраница 1
Введение фенильных групп в метилсиликоны повышает стойкость к окислению, но одновременно повышает температуру замерзания н увеличивает температурный коэффициент вязкости. [1]
При введении фенильной группы в полиметил - и полиэтилсилок-саны повышаются плотность, вязкость и диэлектрическая проницаемость полимеров. Повышаются также температурная область релаксационных явлений, энергии активации вязкого течения, электропроводности и поляризационных явлений. Увеличение числа метпл-фенильных или этилфенильных звеньев в молекуле ведет к более значительному сдвигу области релаксации в сторону высоких температур, росту энергии активации, вязкости, плотности и других свойств. [2]
При введении фенильных групп к атому углерода, заряженному отрицательно, устойчивость карб-анионов возрастает. [3]
Другой метод введения фенильной группы в пиридиновое ядро, приводящий к смеси 2 -, 3 - и 4-замещенных фенил-пиридинов, состоит во взаимодействии пиридина с хлористым фенилдиазо-нием ( стр. Этот метод также может быть с успехом применен для получения смеси фталимидопиридинов, но, очевидно, не может быть применен к гомологам пиридина. [4]
Установлено, что введение фенильной группы повышает ингибиторный эффект 2-меркаптоимидазола, а метильной группы - понижает его. [5]
Установлено, что введение фенильной группы повышает ингибиторный эффект 2-меркаптоимидазола, а метальной группы - понижает его. [6]
Это значит, что введение фенильной группы в парафин или циклопарафин дает молекулу, имеклДую адсорбируемость такого же порядка, как ( а) и ( в); разница же в адсо би-руемости циклопентильной, циклогексильной, н-алкильной и разветвленной алкильной групп относительно невелика. [7]
Значительно большее изменение наблюдается при введении фенильной группы в молекулу уксусной кислоты, поскольку уменьшение значения рК указывает на то, что фенильная группа является значительно более электронопритягивающей ( отрицательной), чем метальная группа. [8]
Аналогично, следует ожидать, что введение фенильной группы к C-I привело бы к большому увеличению скорости реакции зкзо-нор-борннльных субстратов, если бы теория образования неклассйческих ионов была правильной, из-за создания положительного заряда в этом положении в неклассическом промежуточном ионе. [9]
Сравнение энергий активации для этилбензола и дифенилметана показывает, что введение фенильных групп значительно более эффективно, чем введение метальных. [10]
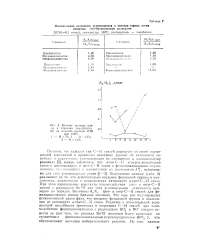 |
Влияние размера цикла и строения заместителя ( R па скорость распада ПТБ. [11] |
Полученные данные ( табл. 2) указывают на то, что действительно введение фенильной группы в цнк-лопентан, циклогекеан н циклододекан активирует а-трет - С - Н связь. При этом парциальные константы взаимодействия трет - и втор - С - Н связей с радикалом Ви О для этих углеводородов отличаются примерно на порядок. Величины kulkd трет - и втор - С - Н связей для фе-нилциклооктана имеют близкие значения. Это еще раз подтверждает установленный ранее факт, что введение фенильной группы в циклоок-тан не активирует а-трет - С - Н связь. Различие в относительной реакционной способности третичных и вторичных С - Н связей при взаимодействии фенилциклоалканов с радикалами R02 и RO следует отнести за счет того, что радикал Ви О является более активным по сравнению с фенилциклоалкильным пероксираднкалом ( RO. J), что обуславливает меньшую избирательность реакции. [12]
Было установлено, что антиокислительные свойства эфиров непредельных фосфиновых кислот повышаются с 1) введением фенильной группы у диэтиловых, диамиловых и дигексиловых эфиров, 2) наличием инденильной группы у диэтиловых и диалли-ловых эфиров, 3) увеличением длины углеводородного радикала ( от С2 до С6) у диаллиловых и дигексиловых эфиров, 4) увеличением длины цепи радикала эфирной группировки ( от С2 до С) у эфиров / 3-бутоксивинилфосфиновой, / 3-фенилвинилфосфиновой и / 3-гексилоксивинилфосфиновой кислот. [13]
На основании всех вышеизложенных фактов мы можем, естественно, прийти к выводу, что степень влияния, оказываемого введением фенильной группы на увеличение вращательной способности активного соединения, в значительной степени обусловливается удаленностью этой группы от активного комплекса. В известных условиях бензольное ядро может оказаться совершенно не действующим, а именно тогда, когда оно расположено слишком далеко от активного комплекса. [14]
РНК - синтетаза phenylamlne фениламин, C8H6NH2 phenylarsenlmide фениларсинимид, C6H5As: NH phenylarsine фениларсин, CeH5AsH2 phenylatlon фенилирование ( введение фенильной группы в органическое соединение в результате реакций замещения или присоединения) phenylazo фенилазо, C H5N2 ( phenylazo) imlno фенилазоимино, C8H5N2N: ( phenylazo) oxy фенилазокси, CeHsN2O phenyl benzoate фенилбензоат, СвН5СООС6Н5 phenyl berate фенилборат, B ( OCeH5) 3 phenylboratlon фенилборирование ( присоединение фенильной группы и борсодержащего остатка по кратной углерод-углеродной связи) phenylbutazone форм, фенилбутазон, CI9H20N2O2 phenyl butyrate фенилбутират, C3H7COOCeH5 phenyl carbarn at f фенилкарбамат, NH2COOC6HS phenylcarbamoyl фенилкарбамоил, C6H5NHCO phenyl carbonate фенилкарбонат, ( C8HtO) 2CO phenyl cellosolve фенилцеллозольв, CaH5OC2H4OH phenylcyanamlde фенилцианамид, C6H5NHCN phenyl cyanate фенилцианат, CeH5OCN phenyl disulfide фенилдисульфид, CeH S2CeH5 phenyldithlo фенилдитио, C6H6SS phenylene фенилен, CeH4 phenylenebls ( aio) фенилендиазо, N2S6H4N2 phenylenedlamlne фенилендиамин, NH2C6H4NH2 phenylenedloxy фенилендиокси, OCeH4O phenylephrine фарм. [15]
Страницы: 1 2 3