Значение - энергия - возбуждение
Cтраница 1
Значение энергии возбуждения для каждой из этих структур может быть оценено на основании энергии связи и других относящихся сюда данных. Фактические значения этих величин весьма сомнительны, однако целесообразно предположить, что по возрастанию этой энергии данные структуры можно расположить в следующем порядке: I, III, II, IV, V. Структуре V, в которой две нормальные ковалентные связи заменены полярными, должна соответствовать энергия возбуждения порядка 9 эв, тогда как структуре IV соответствует энергия лишь немного больше, чем половина этой величины. Может оказаться, что важный вклад в стабилизацию данных структур будут вносить я-электроны системы, поскольку в этом случае становится возможным заменить тригональную гибридизацию волновых функций углеводородных атомов диагональной, что приводит к значительному упрочнению карбонильной связи. В результате этого процесс диссоциации карбонильной группы становится энергетически значительно более предпочтительным. [1]
Вопрос о соответствии значений энергий возбуждения однотипных электронных состояний изоэлектронных двухатомных молекул был рассмотрен Шифриным [463] ( см. стр. [2]
В табл. 4 приведены значения энергий возбуждения для некоторых инертных газов [24, 44], применяющихся в качестве газов-носителей. [3]
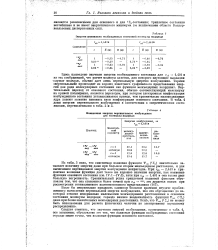 |
Энергии наинизших возбужденных состоянии молекулы водорода. [4] |
Следует отметить, что значения энергий возбуждения, приведенные в табл. 4, все завышены; обусловлено это тем, что волновые функции возбужденных состояний гораздо менее точны, чем волновые функции основного состояния. [5]
Когда в таблицах приведены два и больше значений энергии возбуждения для одной линии, то это объясняется случайным совпадением длин волн линий, соответствующих переходам между термами с разными значениями энергий верхних уровней. Если совпадают линии, принадлежащие ионам разной кратности, то сначала приводится потенциал возбуждения линии иона более низкой степени ионизации. [6]
Остается невыясненным следующий вопрос: в какой степени установленная корреляция значений энергии возбуждения / - полосы, которые рассчитываются в используемой здесь модификации метода Паризера - Парра и по методу Хюккеля, зависит от выбора параметров ( ср. [7]
В предыдущем параграфе было показано, каким образом могут быть найдены значения энергии возбуждения и волновые функции молекул примеси, внедренных в кристаллическую матрицу. В этих расчетах было учтено все кулоновское взаимодействие между зарядами. [8]
Поскольку полосы, соответствующие интеркомбинационным переходам, в спектре NH не наблюдались, значения энергий возбуждения синглетных состояний молекулы NH экспериментально определены не были. Так как в основном состоянии Х32 - молекула NH диссоциирует на атомы в основных состояниях. [9]
Выполненные недавно [ 18516, 2703а ] квантово-механические расчеты молекулы N02 приводят к значениям энергий возбуждения низко лежащих электронных состояний, в основном согласующихся с принятыми в настоящем Справочнике. [10]
Однако при таких сравнительно грубых вычислениях далеко не всегда получается хорошее совпадение со значениями энергии возбуждения молекул, найденными спектроскопическим путем. Для определения этих величин необходимы значительно более точные расчеты. [11]
Для резонансных интегралов p j между соседними атомами углерода использовались их эмпирические значения, что приводит к удовлетворительному согласию вычисленного и экспериментального значений энергии возбуждения для самой длинноволновой полосы в спектре мономеров. Были произведены расчеты теоретических значений резонансных интегралов модифицированным методом Мулликена [6] для я - и 0-ориентации 2 / 7-атомных орбит в простой модели парациклофанов. Оказалось, что для межатомных расстояний, встречающихся в наших расчетах, вычисленные таким способом значения резонансных интегралов дают практически одинаковые величины константы пропорциональности Ktj для любой ориентации 2 / ьатомных орбит. Поэтому мы приняли, что для всех ij выполняется условие P J KStj. Константа К была определена при помощи эмпирического значения р - 2 388 эв для бензола. [12]
Для резонансных интегралов р; между соседними атомами углерода использовались их эмпирические значения, что приводит к удовлетворительному согласию вычисленного и экспериментального значений энергии возбуждения для самой длинноволновой полосы в спектре мономеров. Поэтому мы приняли, что для всех ij выполняется условие PJJ KSij. Константа К была определена при помощи эмпирического значения 3 - 2 388 ев для бензола. [13]
Однако имеются достаточно веские экспериментальные и теоретические доказательства наличия прямого оптического возбуждения СТ-состояний в гомомолекулярных кристаллах, в особенности в более сильно окрашенных членах полиаценового ряда и особенно при значениях энергии возбуждения, меньших их ширины запрещенной зоны. [14]
Если принять во внимание ограничения k kp и k k - f q kp, то становится ясно, что при заданном значении пол-ного / импульса Ид разрешены не все значения энергии возбуждения. [15]
Страницы: 1 2