Ряды - активность
Cтраница 1
Ряды активности окислов и сульфидов металлов различаются. Можно полагать, что сера, содержащаяся в осерненных катализаторах, играет существенную роль в их каталитической активности. [2]
 |
Изменение энергетического профиля при переходе от газовой фазы ( 1 к полярному растворителю. [3] |
Ряды активности нуклеофилов в газовой фазе и в растворе могут быть различны, а также могут меняться в зависимости от растворителя. Например, газофазный ряд F CN Вг Г обращается при переходе к протонным средам, однако в апротонных растворителях ряд нуклеофильности ( CN - Вг Г) такой же, как в газовой фазе. [4]
Ряды активности нуклеофилов в газовой фазе и в растворе Могут быть различны, а также могут меняться в зависимости от Растворителя. Например, газофазный ряд F СГ Вг - Г об-ращается при переходе к протонным средам, однако в апротон - Йых растворителях ряд нуклеофильности ( СГ Вг - 1 -) такой ike, как в газовой фазе. [5]
Ряды активности металлов в реакциях гидрирования этилена при 0 С и дейте-рирования при - 100 С совпадают. Значит, и в случае дейтерирования есть корреляция между активностью катализатора и процентом d - характера интерметаллической связи. [6]
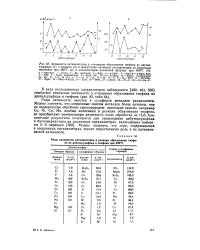
Ряды активности окисных и сульфидных катализаторов в отношении крекинга тиофена близки ( табл. 23, рис. 35), возможно, потому, что активность катализаторов зависит от природы иона - металла. Но не исключено, что окислы металлов в условиях реакции ( даже в опытах, проведенных импульсным методом) частично осерняются и поэтому поверхностный состав сульфидов и так называемых окислов металлов одинаков. Сходные ряды активности сульфидных катализаторов получаются в случае превращения тиофенов различного строения ( см. рис. 35), что может указывать на одинаковый механизм разложения этих соединений. [7]
Однако ряды активности катализаторов, приведенные в разных источниках, не полностью совпадают. Причина в том, что относительная реакционная способность катализаторов зависит в каждом конкретном случае от природы субстрата, реагента и условий проведения реакции. Количество вводимого катализатора колеблется от более, чем эквимольного, до измеряемого несколькими мольными процентами. Алкилиррвание олефиноэ кислотами Льюиса катализируется лишь в присутствии протонодонорного сокатализатора. Особые эффекты обеспечивают суперкислоты [ 612J, , в частности SbFs-HF [613], в среде которых протонируется не только реагент, но и ароматический субстрат. [8]
Рассматривая приведенные выше ряды активностей катализаторов, можно сделать очень важный вывод, что гидрирующая, изоме-ризующая и расщепляющая активности окисных катализаторов ниже активностей сульфидных катализаторов, а расщепляющая активность металлических катализаторов может быть даже выше, чем активность сульфидных катализаторов. Чтобы оценить роль носителя, целесообразно определить удельную активность различных катализаторов - отношение степени превращения веществ в реакциях гидрирования, изомеризации и расщепления к одинаковому числу атомов Мо или W на поверхности катализатора, условно принимая во всех случаях мономолекулярный слой. [9]
Таким образом, ряды активности катализаторов по температурам окисления различных групп органических веществ качественно совпадают. Однако при этом отсутствует корреляция между температурами окисления различных органических веществ для каждого отдельного катализатора. [10]
Рассматривая приведенные выше ряды активностей катализаторов, можно сделать очень важный вывод, что гидрирующая, изоме-ризующая и расщепляющая активности окисных катализаторов ниже активностей сульфидных катализаторов, а расщепляющая активность металлических катализаторов может быть даже выше, чем активность сульфидных катализаторов. Чтобы оценить роль носителя, целесообразно определить удельную активность различных катализаторов - отношение степени превращения веществ в реакциях гидрирования, изомеризации и расщепления к одинаковому числу атомов Мо или W на поверхности катализатора, условно принимая во всех случаях мономолекулярный слой. [11]
 |
Зависимость lg / ( реак-ции элиминирования серы из диэтилсульфида при Г 500 С от числа rf - электронов. [12] |
Таким образом, наиболее хорошо наблюдаемые ряды активности катализаторов объясняются в предположении протекания реакции через стадию образования поверхностного комплекса с переносом заряда от атома серы к катиону. [13]
На основе своих исследований Боресков [94] и несколько ранее Андерсон с сотрудниками [96] составили ряды активности окислов в отношении процесса полного окисления метана, из которых следует, что наиболее активны окислы переходных металлов. [14]
В тех исследованиях, где сопоставляются результаты, полученные для различных катионных форм цеолита, выявлены ряды активности, похожие на те, которые ранее приводились для аналогичных цеолитов в реакциях углеводородов, протекающих по карбониевоион-ному механизму. Например, по активности в дегидратации этанола при 310 С [23] катионные формы цеолита X, морденита и цеолита А располагаются в такой последовательности: CaNaLi-MgK-Ba, MgBaCaNaLiK и MgCaLiNaK. Активность К-формы цеолитов ничтожно мала. [15]
Страницы: 1 2 3