Скорость - реакция - поликонденсация
Cтраница 3
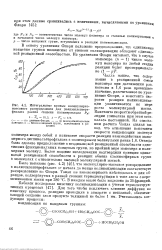
На рис. 42 показано, как изменяется скорость реакции поликонденсации си-аминоэнантовой кислоты в твердой и расплавленной фазах в присутствии раз личных количеств окиси магния. [31]
В случае достаточно быстрых необратимых процессов, когда скорость реакции поликонденсации сравнима со скоростью дозировки реагентов, картина меняется и появляется возможность получения высокомолекулярного продукта при неэквимолярном соотношении реагентов ( см. стр. [32]
Некоторые исследователи [23-25] рассматривают модели, в которых константа скорости реакции поликонденсации зависит т длины реагирующих молекул. В работе [26] обращено внимание на то, что при расчетах кинетики поликонденсации необходимо учитывать контракцию системы в ходе процесса. Возникающая при неучете этого ошибка может, по мнению авторов [26], достигать нескольких десятков процентов. [33]
В промышленности успешно используются фенольные смеси, компоненты которых крайне различны по скорости реакции поликонденсации. [34]
В практике этим пользуются редко из-за повышения вязкости системы и соответстт вующего понижения скорости реакции поликонденсации. [35]
 |
Влияние скорости нагрева на групповой химический состав ЖНП. / - растворимые фракции. 2 - кар-боиды. 3 - карбены. 4 - мапьтены. 5 - асфальтены. [36] |
Уменьшение вязкости пластической массы при повышении скорости нагрева объясняется преобладанием скорости процессов деструкции над скоростью реакции поликонденсации, в результате чего происходит накопление жидко-подвижных веществ в пластической массе. Интенсивность нагрева спекающихся углей влияет на выход и качество ЖНП в областях скоростей, лежащих за пределами и характерных для процесса слоевого коксования. [37]
При повышении температуры коксования скорость испарения и реакций распада составляющих нефтяных остатков увеличивается быстрее, чем скорость реакций поликонденсации, вследствие различной энергии активации реакций. Разрыв во времени между реакциями распада и конденсации способствует выносу из зоны реакций некоторой доли накапливающихся на поверхности частиц структурных звеньев распавшихся молекул и снижает в конечном счете выход кокса. При пониженных давлениях в паровой фазе процесс конденсации структурных звеньев может протекать ограниченно. [38]
Скорость реакций поликонденсации, протекающих по гемолитическому механизму, как правило, слабо зависит от природы растворителя, тогда как скорость реакций поликонденсации гетероли-тического типа ( особенно ионных) в полярных растворителях увеличивается. Однако при этом следует помнить, что молекулярный вес образующегося полимера зависит не столько от абсолютного значения скорости реакций роста, сколько от соотношения между скоростями реакций роста и обрыва. Поэтому влияние природы растворителя на величину молекулярного веса полимера может быть неоднозначно. [39]
 |
Интегральные кривые молекулярно-массового распределения для полиэтилентере-фталата разной степени полимеризации Рп. [40] |
Чалла нашел, что содержание в реакционной смеси мономера при достижении равновесия в 1 6 раза превышает значение, рассчитанное по уравнению Флори, а константа скорости реакции поликонденсации увеличивается по мере роста молекулярной массы. [41]
Так как саморегулирование обусловлено соизмеримостью скорости реакции роста цепи полимера и скорости дозирования одного из мономеров, то, изучая зависимость молекулярного веса от скорости дозирования, можно определить величину скорости реакции поликонденсации, что сделать прямыми методами часто бывает затруднительно. [42]
С учетом того, что производство смол складывается из процесса поликонденсации и побочных реакций полимеризации и дегидратации, вязкость конечных продуктов зависит от скоростей протекания всех реакций, а остаточная кислотность - только от скорости реакций поликонденсации, автором с сотрудниками была предпринята попытка найти дополнительный критерий для определения оптимального момента остановки технологического процесса. Этот критерий должен характеризовать структурные изменения продуктов, которые в неявной форме влияют на результаты аналитического контроля. [43]
Поскольку энергии активации отдельных реакций термолиза различаются между собой весьма существенно, то температура как параметр управления процессом позволяет обеспечить не только требуемую скорость термолиза, я прежде всего регулировать соотношение между скоростями распада и уплотнения и, что особенно важно, между скоростями реакций поликонденсации, тем самым свойства фаз и условия кристаллизации мезофазы. При этом регулированием продолжительности термолиза представляется возможным обрывать на требуемой стадии химическую эволюцию в зависимости от целевого назначения процесса. С позиций получения кокса с лучшей упорядоченностью структуры коксование сырья целесообразно проводить при оптимальной температуре. [44]
Поскольку энергии активации отдельных реакций термолиза различаются между собой весьма существенно, то температура как параметр управления процессом позволяет обеспечить не только требуемую скорость термолиза, но и регулировать соотношение между скоростями распада и уплотнения, а также, что особенно важно, между скоростями реакций поликонденсации, тем самым меняя свойства фаз и условия кристаллизации мезофазы. При этом регулированием продолжительности термолиза представляется возможным обрывать на требуемой стадии химическую эволюцию в зависимости от целевого назначения процесса. Для получения кокса с лучшей упорядоченностью структуры коксования сырья целесообразно проводить при оптимальной температуре. При пониженных температурах из-за малой скорости реакций деструкции в продуктах термолиза будут преобладать нафтено-ароматические структуры с короткими алкильными цепями, которые препятствуют дальнейшим реакциям уплотнения и формированию мезофазы. При температурах выше оптимальной скорости реакций деструкции и поликонденсации резко возрастают. Вследствие мгновенного образования большого числа центров кристаллизации коксующийся слой быстро теряет пластичность, в результате чего образуется дисперсная система с преобладанием мелких кристаллов. Возникающие при этом сшивки и связи между соседними кристаллами затрудняют перемещение и рост ароматических структур. Более упорядоченная структура кокса получается при средних ( оптимальных) температурах коксования ( - 480 С), когда скорости реакций деструкции и уплотнения соизмеримы со скоростью роста мезофазы. Коксующийся слой при этом более длительное время остается пластичным, что способствует формированию крупных сфер меэофазы и более совершенных кристаллитов кокса. [45]
Страницы: 1 2 3 4