Скорость - первая стадия - реакция
Cтраница 1
Скорость первой стадии реакции ( образование ангидрида) пропорциональна квадрату концентрации кислоты. Эта стадия является медленной по сравнению со следующей и, следовательно, определяющей скорость. Для второй стадии реакции еще не сформулирован механизм, который позволил бы объяснить тот факт, что расщепление ангидрида происходит легко лишь в кислой среде. В нейтральной и щелочной средах пирофосфорная кислота чрезвычайно устойчива, а алкоголиз ее значительно замедляется в присутствии слабого органического основания. [1]
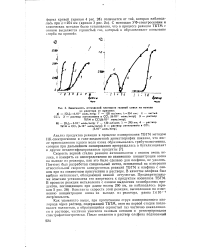 |
Зависимость оптической плотности газовой смеси на выходе. [2] |
Скорость первой стадии реакции антиозонантов с озоном очень велика, и измерить ее непосредственно по изменению концентрации озона на выходе из реактора, как это было сделано для олефина, не удалось. Поэтому был разработан специальный метод, основанный на измерении относительной скорости конкурентных реакций ТБТМ и олефина с озоном при их совместном присутствии в растворе. В качестве олефина был выбран метилолеат, обладающий низкой летучестью. Предварительными опытами установлена его инертность к продуктам озонолиза ТБТМ. В процессе реакции метилолеата с озоном выделения газообразных продуктов, поглощающих при длине волны 290 нм, не наблюдалось ( кривая 5 рис. 26) - Константа скорости этой реакции, вычисленная по изменению концентрации озона на выходе, из реактора, равна lXlOh6 литр / моль-сек. [3]
Взяты константы скорости первой стадии реакции. [4]
Оказалось, что скорость первой стадии реакции высока и имеет второй порядок: первый по кетону и первый по димеру реактива Гриньяра. Однако после того, как половина магнийоргани-ческого соединения прореагирует, скорость реакции становится значительно ниже первоначальной. [5]
Здесь k C - скорость первой стадии реакции, зависящая только от температуры и от концентрации исходного вещества С, которая полагается постоянной; а - коэффициент теплоотдачи в окружающую среду; S и и - соответственно поверхность и объем реакционного сосуда. [6]
Здесь kiC - - скорость первой стадии реакции, зависящая только от температуры и от концентрации исходного вещества С, которая полагается постоянной; а - коэффициент теплоотдачи в окружающую среду; S и со - соответственно поверхность и объем реакционного сосуда. [7]
В наибольшей степени от характера среды зависит скорость первой стадии реакции типа SN ], поскольку при этом происходит разделение разноименных зарядов. Поэтому гетеролиз нейтральных субстратов типа Е - Y, не обладающих цвиттер-ионным характером, значительно ускоряется ростом диэлектрической постоянной среды. В газовой фазе и инертных растворителях с низкой диэлектрической постоянной такие соединения гетеролизуются очень медленно. Поэтому дальнейший рост е оказывает второстепенное влияние. [8]
Так, при окислении ионами МпОТ или СгО - скорость первой стадии реакции часто меньше, чем последующих стадий, в которых участвуют ионы марганца и хрома в низших степенях окисления, способные координировать окисляемый лиганд. Сам факт координации, при котором происходит смещение электронной плотности лиганда в сторону льюисовской кислоты должен увеличивать окислительную способность комплексных частиц. Хотя эту гипотезу трудно проверить, но для Си ( II), Tl ( III), Fe ( III) и им подобных окислителей общепринят внутрисферный механизм окисления и есть данные, подтверждающие это. [9]
Причина этого на первый взгляд непонятного явления состоит в том, что под действием катализатора скорость первой стадии реакции сильно возрастает и становится выше скорости второй стадии. В результате общая скорость реакции приближается к скорости стадии дегидратации. Такая смена механизма представляет собой достаточно широко распространенное явление. [10]
Причина этого на первый взгляд непонятного явления состоит в том, что под действием катализатора скорость первой стадии реакции сильно возрастает и становится выше скорости второй стадии. В результате общая скорость реакции приближается к скорости стадии дегидратации. Такая смена механизма представляет собой достаточно широко распространенное явление. [11]
Найденная зависимость хорошо согласуется с наблюдаемой на практике ( см. рис. 8.12 и табл. 8.6); п зависит от природы полимера и скорости первой стадии реакции. [12]
Свидетельствуют ли полученные данные о протекании реакции по двухста-дийному механизму. Почему константа скорости первой стадии реакции в случае X С1 в два раза больше, чем для X Вг, а вторая стадия проходит медленнее. [13]
Предполагаемый механизм взаимодействия реактивов Гриньяра с карбонильными соединениями подтверждается результатами кинетических измерений. Оказалось, что скорость первой стадии реакции высока и имеет первый порядок по ке-тону и второй порядок по димеру реактива Гриньяра. Однако после того как половина магнийорганического соединения вступит в реакцию, скорость реакции становится значительно ниже первоначальной. [14]
На основе разумных допущений относительно величины некоторых квантовомеханических параметров можно сделать заключение, что второй углеродный атом аниона глиоксалина должен нести больший заряд электронов, чем 4 - й или 5 - й атомы. Кажется естественным, что скорость первой стадии реакции должна зависеть от распределения заряда в невозмущенном реагенте. Так, при азосочетании первый переходный комплекс обладает наиболее высокой потенциальной энергией и ее величина, по-видимому, отвечает первоначальному невозмущенному распределению зарядов. При иодировании второй переходный комплекс находится на наиболее высоком энергетическом уровне, совпадающем по высоте с энергетическим уровнем промежуточного соединения. [15]
Страницы: 1 2