Скорость - депротонирование
Cтраница 1
Скорость депротонирования С - Н - кислоты ( к в табл. 2.7.19; к 1 относится к обратной реакции) в определенных условиях можно использовать как меру кинетической кислотности данной кислоты и устойчивости карбаниона, принимая, что между структурой переходного состояния депротонирования и структурой образующегося карбаниона существует близкая аналогия. К реакциям, скорость которых определяется скоростью депротонирования, относятся катализируемое основаниями галогенирование и изотопный обмен водорода. Скорости подобных реакций действительно представляют собой количественную меру кинетической кислотности, если только внутренняя рекомбинация ионных пар, включающих карбанион, в исходную С - Н - кислоту не существенна и на скорость не влияют другие специальные факторы, например пространственные эффекты в переходном состоянии. Иногда кинетическая кислотность является единственным способом оценки устойчивости карбанионов, например в случае очень слабых С - Н - кислот типа бензола или алканов. Обычно для кинетической и термодинамической кислотностей наблюдается линейное соотношение свободных энергий, и поэтому скорость депротонирования можно использовать для предсказания термодинамической кислотности. [1]
Влияние атомов галогенов на скорость депротонирования основанием в 1-арилнитроэтанах довольно значительно. Так, пара-атомы фтора и хлора увеличивают скорость депротонирования в 50 % - ном метаноле в 1 6 и. [2]
При использовании в качестве растворителя ацстоиигрнла скорость депротонирования АгМНз до ArNH2 определяет общую скорость процесса. [3]
Если ионная пара разделяется и быстро распределяется в растворе, так что каждый акт депротонирования приводит к обмену, то полученные данные об обмене являются точным критерием скорости депротонирования. Однако при многих сочетаниях растворителя и основания ионная пара может возвращаться к исходным реагентам со скоростью, превосходя - Шей скорость нротонирования карбаншна растворителем. [4]
Тот факт, что отщепление водорода от промежуточного продукта азосочетания представляет собой стадию, определяющую скорость реакции, был доказан Золлингером на примере дей-терированных компонентов азосочетания, при взаимодействии которых с диазосоединениями в ряде случаев наблюдался значительный кинетический изотопный эффект. Таким образом, скорость депротонирования проявляется кинетически и реакция соответствует усложненному математическому выражению (7.57) ( стр. Согласно этому, основание, отщепляющее ион водорода, играет важную роль как для скорости всей реакции, так и для направления ( орто - или пара -) азосочетания. [5]
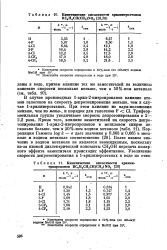 |
Кннетииеские кислотности арилннтроэтанов RC H4CH ( CH3 NO2.| Кинетические кислотности арилни-тропропанов RCeH4CH2CH ( CH3 N02. [6] |
Трифтор-метильная группа увеличивает скорость депротонирования в 2 - 2 2 раза. Более низкое значение в водном метаноле не является неожиданным, цоскольку из-за низкой диэлектрической константы СН3ОН передача полярного эффекта заместителя происходит эффективнее. [7]
Скорость депротонирования С - Н - кислоты ( к в табл. 2.7.19; к 1 относится к обратной реакции) в определенных условиях можно использовать как меру кинетической кислотности данной кислоты и устойчивости карбаниона, принимая, что между структурой переходного состояния депротонирования и структурой образующегося карбаниона существует близкая аналогия. К реакциям, скорость которых определяется скоростью депротонирования, относятся катализируемое основаниями галогенирование и изотопный обмен водорода. Скорости подобных реакций действительно представляют собой количественную меру кинетической кислотности, если только внутренняя рекомбинация ионных пар, включающих карбанион, в исходную С - Н - кислоту не существенна и на скорость не влияют другие специальные факторы, например пространственные эффекты в переходном состоянии. Иногда кинетическая кислотность является единственным способом оценки устойчивости карбанионов, например в случае очень слабых С - Н - кислот типа бензола или алканов. Обычно для кинетической и термодинамической кислотностей наблюдается линейное соотношение свободных энергий, и поэтому скорость депротонирования можно использовать для предсказания термодинамической кислотности. [8]
Влияние атомов галогенов на скорость депротонирования основанием в 1-арилнитроэтанах довольно значительно. Так, пара-атомы фтора и хлора увеличивают скорость депротонирования в 50 % - ном метаноле в 1 6 и. [9]
В общем случае стадией, определяющей скорость реакции, при электрофильном замещении является образование а-комплекса. В исключительных случаях скорость реакции может также определяться скоростью депротонирования а-комплекса. [10]
Это составляет единственное различие двух родственных реакций - отщепления и замещения. Как уже было отмечено ранее, для карбокатионов скорость захвата нуклеофильного агента превышает скорость депротонирования, т.е. 5д1 El, и это соотношение практически не удается изменить заменой растворителя или нуклеофильного агента. Изменение в соотношении продуктов б дД - замещения и Е - элиминирования может быть достигнуто в ионизирующих, полярных и ненуклеофильных растворителях, таких, как трифгорметансульфокислота, и суперкислотах FSOsH-SbFs, SC QF-SbFs или в тех случаях, когда карбокатнонный центр стерически полиостью блокирован разветвленными алкильными группами. [11]
А и В одинаковы для разных углеводородов. Стрейтвизер и др. [19] показали, что в ряде случаев, для которых имеются необходимые данные по равновесиям, это соотношение действительно выполняется; оно было использовано для оценки кислот-ностей углеводородов, когда удается измерить скорости депротонирования, но не равновесия. [12]
 |
Кривые ток - потенциал при окисления ароматического амииа в присутствии кислоты. [13] |
В ацетонитрилс, где депротоиироваиие идет сравнительно медленно, получаемые кривые [33] обычно сходны с кривыми, приведенными на рис. 2.19 о Депротоиироваиие ArNH протекает так медленно, что окисляются только уже присутствующие свободные молекулы ArNH2, и предельный ток уменьшается пропорционально количеству добавленной кислоты. При добавлении воды скорость денротоиирования увеличивается, и предельный ток оказывается истинно кинетическим: он определяется константами скорости ИГР и йобГ и концентрацией кислоты ( см. рис 2.196) Наконец, в водно-органических средах с большим содержанием воды скорость депротонирования так велика, что не удается наблюдать заметного уменьшения предельного тока ( см рис. 2.19 б), и при увеличении концентрации кислоты наблюдается лишь сдвиг всей волны к положительным потенциалам. [14]
Многообещающими являются, по-видимому, такие комбинации, как твердый грег-бутоксид калия / краун-эфир / лодходящий растворитель. Рассмотрим вначале некоторые лучше изученные гомогенные системы. Известно, что скорость депротонирования грег-бутоксидом калия зависит как от его концентрации, так и от типа растворителя. Сложность этой системы объясняется. [15]
Страницы: 1 2