Скорость - десульфирование
Cтраница 1
Скорость десульфирования возрастает по мере разбавления реакционной массы водой, образующейся при сульфировании. [1]
Скорость десульфирования обоих катионитов во всех исследованных средах является возрастающей функцией температуры. [2]
Принимая в качестве меры скорости десульфирования величину абсолютной потери емкости ДЕ, представим на рис. 1 зависимость lg A. [3]
Изучение кинетики показало, что при определенных условиях скорость десульфирования не зависит от концентрации сульфокисло-ты и от природы аниона неорганической кислоты; она пропорциональна активности ионов водорода в растворе. [4]
Изучение кинетики показало, что при определенных условиях скорость десульфирования не зависит от концентрации сульфокисло-ты ц от природы аниона неорганической кислоты; она пропорциональна активности ионов водорода в растворе. [5]
Изучение кинетики показало, что при определенных условиях скорость десульфирования не зависит от концентрации сульфокислоты и природы аниона неорганической кислоты; она пропорциональна активности ионов водорода в растворе. [6]
Из приведенных данных следует, что о-алкильные заместители значительно увеличивают скорость десульфирования. По-видимому, наилучшие результаты в температурном интервале 190 - 220 С дает фосфорная кислота или ее 30 % - ный водный раствор при работе-в автоклаве. Используются также и другие минеральнйе кислоты, но наиболее часто серная кислота. Единственное затруднение при работе с серной кислотой состоит в том, что она не только является донором протонов, но может также окислять органические вещества. [7]
Скорость прямой реакции должна возрастать с увеличением концентрации сульфирующего агента, а скорость десульфирования - с увеличением содержания воды в реакционной массе. Скорость сульфирования максимальна в начале процесса и падает к его завершению, в то время как скорость десульфирования начинает играть заметную роль только в конце реакции. По-видимому, приведенные выше значения величин л сульфирования примерно соответствуют тому моменту реакции, когда скоростй ульфироаа7 ния и десульфирования сближаются и устанавливается равновесие. Так как состав продуктов сульфирования в состоянии равновесия - при термодинамическом контроле реакции - может сильно отличаться от первоначального, полученного при кинетическом контроле ( см. 2.1), обратимость реакции сульфирования имеет большое практическое значение. Варьируя концентрацию сульфирующего агента и температуру, во многих случаях удается регулировать скорости прямой и обратной реакций и таким образом направлять течение сульфирования в сторону целевого продукта необходимого строения. [8]
С другой стороны, скорость прямой реакции должна возрастать с увеличением концентрации сульфирующего агента, а скорость десульфирования - с увеличением содержания воды в реакционной массе. Поэтому сместить приведенное выше равновесие вправо можно, увеличивая количество и концентрацию серной кислоты, взятой для сульфирования, или же удаляя воду из реакционной массы. В связи с изложенным количество сульфирующего агента, необходимого для проведения реакции, рассчитывают не по стехиометрическому соотношению, а по концентрации серной кислоты, которая должна получиться после завершения процесса сульфирования. Эта эмпирически найденная оптимальная концентрация отработанной серной кислоты различна для различных соединений и зависит от их реакционной способности: чем легче ароматическое соединение вступает в реакцию сульфирования, тем при прочих равных условиях она может быть ниже. [9]
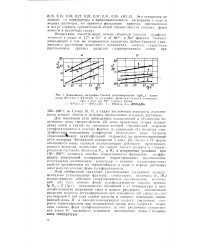 |
Зависимость логарифма степени десульфирования ( gR и катио-нитов КУ-2 ( а и КУ-23 ( б от логарифма продолжительности нагревания. [10] |
J поляризующему действию гид рати рованных нротивоионов; постоянный для всех ионных форм сульфокатионитов наклон прямых lg RK-F ( Igt): независимость отношений i от температуры и продолжительности нагревания смол в водных растворах; значительное снижение скорости десульфирования при повышении pil среды за счет растворения в ней соли слабой кислоты и сильного основания, связывающей ионы II1 в малодиссоцинрующую кислоту; увеличение скорости термогидролиза при подкисленйи среды; рост скорости гидролиза солевых форм сульфокатионитов за счет увеличения концентрации ионов Н, вызванного усилением диссоциации воды с повышением температуры. [11]
Многие противоионы действуют как катализаторы процесса гидролиза и других стадий разложения. Показано, что скорость десульфирования - 8О3Н - групп фенолформальдегидных катионообменников в ионных формах щелочных металлов повышается в ряду Cs Rb К Na Li, т.е. с увеличением эффективного радиуса гидратированного противоиона. [12]
Как сообщалось ранее, - положения нафталина активнее р-положений и соотношение скоростей образования 1 - и 2-сульфо-кислоты меняется от 5 9 до 4 1 с увеличением концентрации серной кислоты от 75 до 95 % при 25 С. Примерно в таком же отношении находятся и скорости десульфирования этих сульфокислот. [13]
Скорость прямой реакции должна возрастать с увеличением концентрации сульфирующего агента, а скорость десульфирования - с увеличением содержания воды в реакционной массе. Скорость сульфирования максимальна в начале процесса и падает к его завершению, в то время как скорость десульфирования начинает играть заметную роль только в конце реакции. По-видимому, приведенные выше значения величин л сульфирования примерно соответствуют тому моменту реакции, когда скоростй ульфироаа7 ния и десульфирования сближаются и устанавливается равновесие. Так как состав продуктов сульфирования в состоянии равновесия - при термодинамическом контроле реакции - может сильно отличаться от первоначального, полученного при кинетическом контроле ( см. 2.1), обратимость реакции сульфирования имеет большое практическое значение. Варьируя концентрацию сульфирующего агента и температуру, во многих случаях удается регулировать скорости прямой и обратной реакций и таким образом направлять течение сульфирования в сторону целевого продукта необходимого строения. [14]
Скорость ее возрастает с увеличением концентрации кислоты. Установлено, что при повышении температуры на 10 С скорость гидролиза возрастает в 2 5 - 3 5 раза. Скорость десульфирования зависит также от природы заместителя в молекуле арилсульфокислот. [15]
Страницы: 1 2