Скорость - десульфирование
Cтраница 2
Электронодонорный заместитель - гидроксильная группа - в большей степени активирует замещение в орто-положение ( k0 kn), и при низкой температуре и кинетическом контроле в суль-фомассе преобладает о-гидроксибензолсульфокислота. По мере течения реакции, особенно при повышении температуры до 100 С, концентрация о-изомера уменьшается и накапливается га-изомер, становящийся при термодинамическом контроле главной составной частью продуктов моносульфирования. Объясняется это тем, что скорость десульфирования о-изомера больше, чем n - изомера. Такого рода изомеризация сульфокислот широко используется в синтезе промежуточных продуктов, особенно нафталинового ряда. [16]
Как видно, продуктами моносульфирования нафталина могут являться а - и р-сульфокислоты. По причинам, о которых сообщалось ранее, сс-положения нафталина более активны, чем р -, и скорость образования ос-сульфокислоты больше, чем скорость образования р-изомера. В таком же отношении находятся и скорости десульфирования этих сульфокислот. Поэтому, создавая такие условия, в которых первично образующаяся а-сульфокислота не успевала бы гидролизоваться, можно получить этот продукт с удовлетворительным выходом. Наоборот, в условиях, благоприятствующих гидролитическим процессам, в сульфомассе накапливается р-изомер, становящийся главным продуктом реакции. Обычно сульфирование нафталина на а-сульфокислоту ведут моногидратом при 60 С. При этом в сульфомассе содержится до 20 % Р - изомера. Фирц-Давидом описано также сульфирование нафталина моногидратом при 0 С. [17]
Продуктами моносульфирования нафталина могут являться 1-и 2 - нафталинсульфокислоты. По причинам, которые обсуждались ранее ( см. 2.3), а-положения нафталина значительно активнее р-положений и соотношение скоростей образования 1 - и 2-сульфо-кислот составляет от 6 до 3 5 - 4, в зависимости от условий проведения реакции. По-видимому, в таком же соотношении находятся и скорости десульфирования этих сульфокислот. Поэтому при кинетическом контроле реакции, который создается максимальным замедлением гидролитических процессов - малое содержание воды в сульфомассе, низкая температура, короткая выдержка - удается получить 1-нафталинсульфокислоту, с удовлетворительным выходом. Наоборот, для преимущественного получения 2-нафталинсуль-фокислоты, необходим термодинамический контроль, создающийся в условиях, благоприятствующих установлению термодинамического равновесия сульфирования и десульфирования, при котором должно уменьшаться содержание 1-сульфокислоты в пользу более стабильного 2-изомера. [18]
Продуктами моносульфирования нафталина могут являться 1-и 2 - нафталинсульфокислоты. По причинам, которые обсуждались ранее ( см. 2.3), а-положения нафталина значительно активнее р-положений и соотношение скоростей образования 1 - и 2-сульфо-кислот составляет от 6 до 3 5 - 4, в зависимости от условий проведения реакции. По-видимому, в таком же соотношении находятся и скорости десульфирования этих сульфокислот. Поэтому при кинетическом контроле реакции, который создается максимальным замедлением гидролитических процессов - малое содержание воды в-сульфомассе, низкая температура, короткая выдержка - удается получить 1-нафталинсульфокислоту с удовлетворительным выходом. Наоборот, для преимущественного получения 2-нафталинсуль-фокислоты, необходим термодинамический контроль, создающийся в условиях, благоприятствующих установлению термодинамического равновесия сульфирования и десульфирования, при котором должно уменьшаться содержание 1-сульфокислоты в пользу более стабильного 2-изомера. [19]
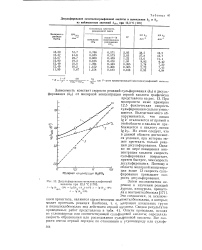 |
Десульфирование метиленсульфоновой кислоты при 12 3 С. [20] |
При молярности ниже примерно 12 5 фактическая скорость сульфирования сильно уменьшается. Из этого следует, что в данной области достигаются условия, при которых может протекать только реакция десульфирования. Однако по мере повышения концентрации кислоты скорость сульфирования возрастает, причем быстрее, чем скорость десульфирования. Поэтому в области молярной концентрации выше 13 скорость сульфирования превышает скорость десульфирования. [21]
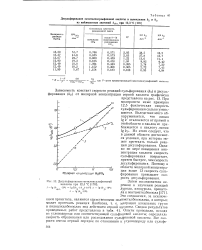 |
Десульфирование метиленсульфоновой кислоты при 12 3 С. [22] |
При молярности ниже примерно 12 5 фактическая скорость сульфирования сильно уменьшается. Из этого следует, что в данной области достигаются условия, при которых может протекать только реакция десульфирования. Однако по мере повышения концентрации кислоты скорость сульфирования возрастает, причем быстрее, чем скорость десульфирования. Поэтому в области молярной концентрации выше 13 скорость сульфирования превышает скорость десульфирования. [23]
Лонг и Поль [204] отметили, что, приняв механизм Коудрея и Дейвиса, нужно предположить, что в 92 - 99 % - ной серной кислоте десульфирование происходит без кислотного катализа. Изучению этого вопроса мешает то обстоятельство, что при таких концентрациях кислоты равновесие полностью сдвинуто в одну сторону; однако в более разбавленной серной кислоте, скажем 10 - 50 % - ной, отчетливо проявляется кислотный катализ этой реакции. Используя данные Крафтса [205] и свои данные, Голд и Сэтчелл [206], а также Лонг и Поль показали, что в 15 - 45 % - ной серной кислоте скорость зависит от функции Гамметта ha, что указывает на участие протона в предкинетическом равновесии. Голд и Сэтчелл отметили, что такая зависимость должна наблюдаться в рамках механизма Коудрея - Дейвиса в том случае, если сульфокислота находится в растворе в основном в виде сульфонат-иона. При концентрации серной кислоты выше 50 % ( lg h0 3) сульфокислота переходит из ионной формы в недиссоциированную, и зависимость от h0 должна исчезать. Используя данные Пинноу [207] и Лантца [208], Голд и Сэтчелл показали, что при концентрации серной кислоты выше 50 % зависимость скорости десульфирования от h0 гораздо менее заметна. Таким образом, почти полностью исчезает кислотный катализ десульфирования. [24]
Все они сводились к построению формальных зависимостей, нередко не имеющих физического смысла. Наличие участков с большой и малой скоростью десульфирования в макропористых образцах катионита КУ-23 ( Н) и подчинение кинетики процесса на каждом участке уравнению реакции первого порядка авторы работы [123] связывают со спецификой пористых катионитов по сравнению с гелевыми катио-нитами. В работе [321] приведены линейные зависимости относительных потерь обменной емкости большого числа сульфокатионитов от продолжительности нагревания в воде ( для начальных участков кинетических кривых) и на этом основании сделан ошибочный вывод о подчинении кинетики десульфирования формальному уравнению реакции первого порядка. [25]
Страницы: 1 2