Полная скорость - реакция
Cтраница 2
Важной особенностью реакций, описываемых приведенной выше схемой, является то, что полная скорость реакции пропорциональна квадратному корню из скорости инициирования. Если же используется гидроперекись, то ее концентрация входит в выражение для скорости реакции в первой степени. Независимым путем было доказано, что в широком интервале условий кинетика распада гидроперекисей соответствует бимолекулярной реакции. [16]
Можно представить гипотетический случаи, когда одна из этих величин достаточно велика, несмотря на то, что полная скорость реакции значительно больше, чем скорость только реакции роста. Справедливость допущения о том, что скорость исчезновения мономера равна скорости реакции роста, подтверждается двумя фактами: во-первых, это допущение приводит к уравнениям, хорошо согласующимся с экспериментальными данными, и, во-вторых, как может быть показано, длина и кинетической, и молекулярной цепи обычно достаточно велика, и рассмотренные выше случаи лишь очень редко соответствуют какой-либо реальной полимеризационной системе. [17]
 |
Влияние размера зерен катализатора на скорость реакции. [18] |
Если скорость процесса на одном из этапов меньше, чем на других, то именно этот этап определяет полную скорость реакции на всех стадиях. [19]
Скорость реакции равна ft [ Ni2 ] [ dto2 - ], и, следовательно, первая ступень определяет полную скорость реакции. [20]
Ре - равновесное давление в системе, то можно видеть, что в условиях, когда определяющей является стадия адсорбции ( и только она), полная скорость реакции пропорциональна отклонению системы от равновесия: Р - Ре. В некоторых случаях скорость может оказаться линейной функцией Р1 / г, если кислород адсорбируется в диссоциированной форме. [21]
Напротив, преимущество теории, изложенной в данной книге, как раз в том и состоит, что в ней неравновесные межфазовые стадии органически входят в соответствующие уравнения; на этой основе получены выражения для полной скорости реакции как функции температуры и парциальных давлений газообразных компонентов системы. [22]
Таким образом, ф2т ] - ( dw / dl), и так как в переходной области ф растет приблизительно до 20, несмотря на то что г ] падает от 1 до / 2, полная скорость реакции dw / dt должна возрасти приблизительно в 200 раз. Из этого следует, как указывают Вейц и Пратер [103], что переходная область между зонами I и II может занять большой температурный интервал. [23]
Как отмечалось выше, простейшее описание кинетики химических реакций дается уравнениями, содержащими только концентрации реагирующих молекул и константы скорости. В состоянии термодинамического-равновесия полные скорости реакций равны нулю, что позволяет установить связь между константами скорости и равновесными концентрациями реагирующих веществ. Отношения последних определяются однозначно-через термодинамические константы равновесия К, величины которых не зависят от механизма реакции и которые выражаются через статистические суммы молекул, участвующих в реакции. Хотя это не вызывает сомнения для равновесных реакций, на любой стадии которых нарушение мак-свелл-больцмановского распределения мало, для неравновесных реакций указанная связь не является строго обоснованной. [24]
Как отмечалось выше, простейшее описание кинетики химических реакций дается уравнениями, содержащими только концентрации реагирующих молекул и константы скорости. В состоянии термодинамического равновесия полные скорости реакций равны нулю, что позволяет установить связь между константами скорости и равновесными концентрациями реагирующих веществ. Отношения последних определяются однозначно через термодинамические константы равновесия К, величины которых но зависят от механизма реакции и которые выражаются через статистические суммы молекул, учпствующих в реакции. Хотя это не вызывает сомнения для равновесных реакций, на любой стадии которых нарушение максвелл-больцмановского распределения мало, для неравновесных реакций указанная связь не является строго обоснованной. [25]
Порядок реакции определяется кинетическим уравнением и чаще всего не совпадает с молекулярностью ( стехиометрией) реакции, так как большинство сложных реакций протекает в несколько стадий. Промежуточные реакции могут оказаться решающими при определении полной скорости реакции. [26]
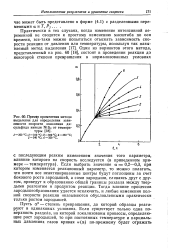 |
Пример применения метода выделения для определения зависимости скорости окисления дисульфида никеля NiS2 от температуры. [27] |
Если выбрать значение ос 0 2 - 0 3, при котором изменяется реакционный параметр, то можно полагать, что почти все неактивированные центры будут поглощены за счет бокового роста зародышей, а сами зародыши, сливаясь друг с другом, приведут к образованию общей границы раздела между твердыми реагентом и продуктом реакции. Тогда влияние процессов зародышеобразования удается исключить, и любые изменения полной скорости реакции оказываются обусловленными практически только ростом зародышей. [28]
Влияние заряда объясняет также и механизм, включающий образование ионной пары, сопровождающееся реакцией превращения внешнесферного комплекса во внутрисферный. Больший отрицательный заряд должен был бы увеличивать концентрацию ионной пары и, следовательно, полную скорость реакции анации. [29]
Следовательно, проникновение в углерод уменьшается. Очевидно, равновесие достигается, когда градиент концентрации возрастет в 3 раза, а глубина проникновения уменьшится в 3 раза и полная скорость реакции ( пропорциональная - кратному расстоянию проникновения) возрастет в 3 раза, где 3 ( в этом примере) является корнем квадратным из коэффициента увеличения удельной константы скорости. [30]
Страницы: 1 2 3