Истинная скорость - химическая реакция
Cтраница 1
Истинная скорость химической реакции представляет собой производную концентрации по времени. [1]
Как выражается средняя и истинная скорость химических реакций. [2]
Чтобы определить истинную скорость химической реакции ( V l), достаточно найти производную концентрации какого-нибудь одного вещества, участвующего в реакции, по времени. [3]
Поэтому для оценки истинной скорости химической реакции в данный момент времени должна быть взята первая производная of концентрации по времени. [4]
Таким образом, интересно исследовать зависимость между истинной скоростью химической реакции и явлением физического транспорта, чтобы определить выражение для полной скорости превращения ( на единицу реакционного объема или на единицу массы катализатора), которое затем можно применять для расчетов реактора по способу, описанному во II и IV главах. [5]
Задаваясь пределом точности, с которой необходимо определять истинную скорость химической реакции, и отвечающим. ДРд / Рл в диффузионном слое, находят соответствующую скорость потоков при данном значении комплекса X. Так, например, если экспериментальная точность определения скорости реакции должна быть более 110 %, то диффузионным сопротивлением можно пренебречь. [6]
Будем считать, что скорость процесса зависит как от истинной скорости химической реакции, так и от скорости переноса массы. [7]
Это дает предпосылки рассматривать зону реакции как проточный реактор с входящими потоками растворяющихся в жидкости компонентов и при построении модели ввести истинную скорость химической реакции, которая определена конкретным атомным механизмом взаимодействия, характерным для гомогенных систем. [8]
Из этой формулы следует, что глубина проникновения вещества в пористый химически активный для него материал определяется отношением скорости его молекулярной диффузии D к истинной скорости химических реакций К. [9]
Если брать изменение концентрации за бесконечно малый промежуток времени, то получим выражение для истинной скорости химической реакции в данный момент времени. [10]
Те же идеи и методы, которые были применены в теории теплового воспламенения для гомогенных реакций, мы применим теперь к вопросу о тепловом режиме гетерогенных экзотермических реакций. Отличие от гомогенных реакций заключается в том, что в этом случае скорость реакции не может уже возрастать неограниченно, вплоть до самых высоких температур. Скорость гетерогенного химического процесса определяется как истинной скоростью химической реакции на поверхности, так и скоростью подвода реагирующих веществ к этой поверхности молекулярной или конвективной диффузией. При низких температурах, пока скорость реакции мала по сравнению со скоростью диффузии ( кинетическая область), суммарная скорость процесса определяется истинной кинетикой на поверхности и экспоненциально возрастает с температурой, согласно закону Аррениуса. Но это возрастание может продолжаться лишь до тех пор, пока скорость реакции не сделается сравнимой со скоростью диффузии. В дальнейшем процесс перейдет в диффузионную область, где скорость его всецело определяется скоростью диффузии и лишь весьма слабо возрастает с температурой. При такой зависимости скорости выделения тепла от температуры и при определенных условиях тепло-отвода возможны три стационарных тепловых режима, из которых средний оказывается неустойчивым, верхний отвечает протеканию реакции в диффузионной, а нижний - в кинетической области. Воспламенение поверхности представляет собой скачкообразный переход от нижнего к верхнему стационарному тепловому режиму. Обратный переход от верхнего теплового режима к нижнему происходит также скачком при критическом условии потухания, не совпадающем с условием воспламенения. [11]
Первая стадия процесса определяется физико-химическими свойствами топливной смеси, а вторая стадия - диффузией и теплообменом. В зависимости от соотношения скоростей двух его стадий различают две предельные области. В первой из них - кинетической - скорость реакции гораздо меньше скорости диффузия реагирующих компонентов, и наблюдаемая скорость процесса совпадает с истинной скоростью химических реакций. Во второй области - диффузионной - скорость процесса всецело определяется переносом вещества в зоне горения, и он лимитируется фактором гидродинамической природы и в первую очередь смещением. [12]
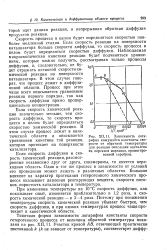 |
Зависимость логарифма кажущейся константы. скорости от обратной температуры для реакции окисления ацетилена на перекиси марганца, промотиро-ванной серебром. [13] |
Наблюдаемая макроскопическая кинетика реакции будет подчиняться уравнениям, которые можно получить, рассматривая только процессы диффузии, и, следовательно, не бу-дет отражать истинной скорости химической реакции на поверхности катализатора. В этом случае говорят, что процесс лежит в диффузионной области. Процесс при этом чаще всего описывается уравнением реакции первого порядка, так как скорость диффузии прямо пропорциональна концентрации. [14]
Последнее обстоятельство говорит о большой кинетической энергии молекул, бомбардирующих стенки сосуда. Молекулярно-кинети-ческая теория, разработанная Клаузиусом, Максвеллом и Б ольцманом, дает возможность рассчитать средние скорости движущихся молекул, число максимальных Столкновений, зависимость этого числа от концентрации молекул, температуры и пр. Все это в одинаковой мере относится не только к газам, но и к растворам, в которых идут химические реакции. Итак, имеется резкое расхождение между общим числом столкновений, которые можно подсчитать, и истинной скоростью химической реакции. [15]
Страницы: 1 2