Случай - параллельная реакция
Cтраница 2
Так же как и в случае параллельных реакций, концентрация целевого продукта на выходе из реактора зависит от двух констант, причем влияние отравления катализатора на эти константы может быть различным. Это усложняет решение задач. [16]
Практически чаще всего приходится встречаться с двумя случаями параллельных реакций. [17]
Таким образом, производная производительности реактора по нагрузке, так же как и в случае параллельных реакций, может быть выражена через концентрации продуктов В и С на выходе из реактора, и, следовательно, при оптимальном распределении нагрузок функции F ( xBi Xa) для всех параллельных реакторов должны принимать одинаковые значения. [18]
Для определения отношений констант скоростей xj можно воспользоваться тем же приемом, что и в случае параллельных реакций, а именно, провести реакцию при недостатке вещества В. [19]
Для определения отношений констант скорости х, можно воспользоваться тем же приемом, что и в случае параллельных реакций, а именно: провести реакцию при недостатке вещества В. [20]
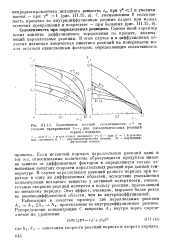 |
Зависимость полной селективности s от степени превращения 1 - ct для последовательных реакций. [21] |
Если истинный порядок параллельных реакций один и тот же, относительные количества образующихся продуктов никак не зависят от диффузионных факторов и определяются только отношением констант скорости параллельных реакций при данной температуре. В случае параллельных реакций разного порядка при переходе в одну из диффузионных областей, вследствие уменьшения концентрации исходного вещества у активной поверхности, относительные скорости реакций меняются в пользу реакции, протекающей по меньшему порядку. Этот эффект, очевидно, выражен более резко во внешнедиффузионной области, чем во внутридиффузионной. [22]
Второй раздел химической кибернетики, занимающийся разысканием оптимальных условий проведения химического процесса, широко использует как классические методы вариационного исчисления, так и новейшие достижения современной математики-динамическое программирование и принцип максимума. В качестве простейшего примера можно указать уже упоминавшийся выше случай параллельных реакций с разными энергиями активации. При осуществлении подобного процесса в каталитическом реакторе идеального вытеснения выгодно повышать температуру катализатора вдоль слоя по мере выгорания исходного вещества. [23]
Второй раздел химической кибернетики, занимающийся разысканием оптимальных условий проведения химического процесса, широко использует как классические методы вариационного исчисления, так и новейшие достижения современной математики - динамическое программирование и принцип максимума. В качестве простейшего примера можно указать уже упоминавшийся выше случай параллельных реакций с разными энергиями активации. При осуществлении подобного процесса в каталитическом реакторе идеального вытеснения выгодно повышать температуру катализатора вдоль слоя по мере выгорания исходного вещества. [24]
Вследствие этой разницы отношение сумм состояний в случае обменной реакции получается в 10 раз больше, чем в случае параллельной реакции. Физический смысл низкой частоты деформационного колебания комплекса D - Вг - Н и, соответственно, высокого значения суммы состояний заключается в значительной вероятности образования активированного состояния, которое может образоваться даже в том случае, если атом дейтерия приближается к молекуле под значительным углом к ее оси. В то же время для образования комплекса D - Н - Вг атом дейтерия должен приближаться к молекуле НВг вдоль ее оси. [25]
При проведении параллельных реакций температуру не всегда выгодно увеличивать, так как это может привести к преобладанию скорости побочной реакции. Поэтому в случае параллельных реакций температуру следует увеличивать только тогда, когда это приводит к преимущественному увеличению скорости основной реакции. [26]
Совсем иной характер носит влияние диффузионного торможения на процесс, включающий параллельные реакции. В этом случае и в диффузионных областях истинная химическая кинетика реакций на поверхности остается фактором, определяющим селективность процесса. Если истинный порядок параллельных реакций один и тот же, относительные количества образующихся продуктов никак не зависят от диффузионных факторов и определяются только отношением констант скорости параллельных реакций при данной температуре. В случае параллельных реакций разного порядка при переходе в одну из диффузионных областей, вследствие уменьшения концентрации исходного вещества у активной поверхности, относительные скорости реакций меняются в пользу реакции, протекающей по меньшему порядку. Этот эффект, очевидно, выражен более резко во внеш-недиффузионной области, чем во внутридиффузионной. [27]
Иной характер носит влияние диффузии реагентов в зерне катализатора на параллельные реакции. В этом случае и в диффузионной области истинная химическая кинетика реакций на поверхности может остаться единственным фактором, определяющим селективность процесса. Если порядок параллельных реакций один и тот же, то селективность процесса не будет зависеть от диффузионных факторов и определяется только отношением констант скорости параллельных реакций при данной температуре. В случае параллельных реакций разного порядка при переходе в диффузионную область, вследствие уменьшения концентрации исходного реагента у активной поверхности, относительные скорости реакций меняются в пользу реакции, протекающей по меньшему порядку. [28]
Если бы величина & - составляла Ю4 ( k - 102), то А и В очень быстро превратились бы в X и Y. Еще в конце раздела 1.5.6.1 указывалось, что только при эндотермических реакциях всегда быстрее всего - образуются наиболее стабильные продукты. В разделе 1.5.8.3 было показано, что в случае параллельных реакций быстрее могут образовываться и термодинамически менее стабильные соединения. [29]
В основе теории параллельных реакций лежит принцип независимого их протекания, согласно которому каждая нз параллельных реакций протекает независимо от всех других электродных реакций, идущих на том же электроде. Из этого принципа следует, что при заданном потенциале и прочих равных условиях скорость какой-нибудь реакции, протекающей одновременно с другими, равна скорости этой же реакции в отсутствие параллельных реакций. Для количественного рассмотрения вопроса введем допущение о том, что электрод под током имеет во всех точках один и тот же потенциал и, следовательно, ток распределяется по всей поверхности равномерно. Поскольку направленное протекание процесса возможно только тогда, когда потенциал под током лежит отрицательнее равновесного потенциала электродной реакции для катодного процесса и положительнее - для анодного процесса, то в случае параллельных реакций это же условие должно соблюдаться для всех реакций. [30]
Страницы: 1 2