Соотношение - интенсивность - пик
Cтраница 1
Соотношение интенсивностей пиков дает возможность рассчитать содержание структур: винил - винил, винил - винили-ден и винилиден - винилиден. [1]
Значение соотношения интенсивностей пиков ионов ( М - ОН) и ( М - Н2О) при 50 зВ для соединения V равно 1 2, а для сульфона IV - 0 5, и при уменьшении энергии электронов оно составляет для рассматриваемых соединений величину 0 3, соответствующую о-метил-замещенному дифенилсульфону. Таким образом, при низких энергиях ионизации специфичность распада молекулярных ионов, связанная с изменением веса замещающей группы и ее природы, не наблюдается. Примечательной особенностью последующего распада образовавшихся ионов является р-разрыв заместителя с отщеплением метального ( для соединения V) или фенильного ( для VI) радикалов. Кроме того, для диссоциативной ионизации этилдифенилсульфона характерно, впервые отмеченное нами, образование иона ( М-2 Н2О), обусловленное отрывом двух молекул воды, сопровождаемое метастабильным переходом. [2]
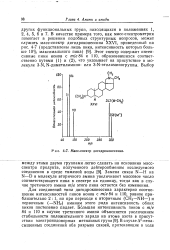 |
Масс-спектр дигидроконессина. [3] |
Для соединений типа дигидроконессина характерно соотношение интенсивностей пиков ионов с т / е 84 и ПО, равное приблизительно 2: 1, но при переходе к вторичным ( СН3 - NH -) и первичным ( - NH2) аминам этого ряда интенсивность обоих пиков постепенно падает. [4]
При оценке СК рентгенографическим методом используют соотношение интенсивностей пиков на дифрактограмме, обусловленных рассеянием лучей кристаллическими областями, и размытого аморфного гало. Однако такую характеристику кристалличности следует считать условной, поскольку в целлюлозе нет строгого разделения на две фазы. В действительности существуют переходные зоны между кристаллическими и аморфными участками, а также дефекты кристаллической решетки и пара-кристаллическая часть. Кроме того, в кристаллической части возможно присутствие разных полиморфных модификаций целлюлозы. Определяемая рентгенографически СК целлюлозы характеризует долю макромолекул, упорядоченных с образованием трехмерной кристаллической решетки, и долю остальных менее упорядоченных макромолекул. [5]
Определяемые соединения идентифицируют по времени удерживания в хроматографической колонке и соотношению интенсивностей пиков характеристических ионов с соответствующими значениями для стандартных смесей. Концентрацию ОХДФ определяют по меченому ОХДД. В связи с тем, что изотопно меченый 1 2 3 7 8 9 - ГкХДЦ используется в качестве внутреннего стандарта, он не вводится в анализируемый образец перед экстракцией и не может быть использован для определения природного аналога. Последний определяют по среднему значению сигнала для других изотопно меченых гексахлордибензодиоксинов. Качество анализа обеспечивается калибровкой прибора, контроля за операциями выделения и очистки соединений. [6]
Определяемые соединения идентифицируют по времени удерживания в хроматографической колонке и соотношению интенсивностей пиков характеристических ионов с соответствующими значениями для стандартных смесей. Концентрацию ОХДФ определяют по меченому ОХДД. В связи с тем, что изотопно меченый 1 2 3 7 8 9 - ГкХДД используется в качестве внутреннего стандарта, он не вводится в анализируемый образец перед экстракцией и не может быть использован для определения природного аналога. Последний определяют по среднему значению сигнала для других изотопно меченых гексахлордибензодиоксинов. Качество анализа обеспечивается калибровкой прибора, контроля за операциями выделения и очистки соединений. [7]
Состав и координация атомов кремния и кислорода в слое определяются из соотношения интенсивности пиков Si2p и Ols в спектре и химических сдвигов А. Менее определенны данные о ее строении. [8]
Как видно из таблицы, в масс-спектрах соединений первой и второй групп соотношение интенсивностей пика арилметиленового катиона к молекулярному ( [ М - Н ] ДМ ]) уменьшается с понижением ионизирующего напряжения. Поведение п-метоксито-луола при взаимодействии с электронами несколько отличается от выше рассмотренных соединений. Это объясняется, очевидно, высокой стабильностью / г-метокситолил-катиона, а также тем, что под влиянием сильно донорной групп ОСН3 потенциал ионизации радикала НзСО - - РН-СН2 значительно уменьшается. Его малая интенсивность обусловлена, по-видимому, крайней нестабильностью М 2, вследствие чего он подвергается быстрому распаду. [9]
По-видимому, вероятность локализации заряда на одном из двух ароматических ядер определяется наличием или отсутствием связи бензильного заместителя с атомом азота гетероциклического ядра, при этом соотношение интенсивностей пиков ионов с массой ( М - 77) и 91 довольно постоянно для каждого из рассмотренных типов замещения: оно меньше 1 для Л / - бензил-производных и лежит в пределах 10 для С-бензилпроизводных. [10]
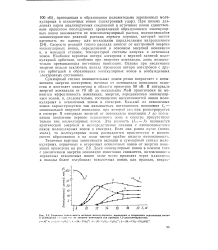 |
Типичная зависимость вкладов молекулярного, первичных и вторичных осколочных ионов в суммарный по шый тон от энергии ионизации ( на примере 1 2-диэтилгидразина. [11] |
В интервале энергий ионизации от 70 эВ до нескольких МэВ практически не меняются эффективность ионизации, энергия, передаваемая молекулярным ионам, и, следовательно, соотношение интенсивностей пиков молекулярных и осколочных ионов в спектрах. Каждый из осколочных ионов характеризуется так называемым потенциалом появления / п - минимальной энергией ионизации, при которой его пик регистрируется в спектре. В интервале энергий от потенциала ионизации / до потенциала появления первого осколочного нона в спектре присутствует только пик молекулярных ионов. Эта разность ( / i - /) называется критической энергией и непосредственно связана с интенсивностямп пиков молекулярных ионов в спектрах. Если она равна нулю ( очень мала), то молекулярные ионы распадаются практически в момент своего образования и их пики имеют крайне низкую интенсивность. [12]
Важное условие работы прибора - быстрая запись масс-спектра, к-рый должен регистрироваться за время, гораздо меньшее, чем время выхода хроматографич. Медленная запись масс-спектра может исказить соотношение интенсивностей пиков в нем. Скорость регистрации масс-спектра ( скорость сканирования) определяется масс-анализа-тором. Наименьшее время сканирования полного масс-спектра ( неск. ЭВМ, построение хроматограмм и обработка масс-спектров производится автоматически. Через равные промежутки времени по мере элюирования компонентов смеси регистрируются масс-спектры, количеств, характеристики к-рых накапливаются в памяти ЭВМ. Для каждого сканирования производится сложение интенсивностей всех регистрируемых ионов. Задавая номер сканирования, можно вызвать из памяти масс-спектр в любой точке хроматограммы. [13]
Графический спектр позволяет с легкостью оценить соотношения интенсивностей пиков, что более важно для целей идентификации, нежели абсолютные значения интенсивностей пиков. Кроме того, можно быстро сделать общие выводы о характере анализируемого спектра. [14]
Экспериментальный спектр 1-бром - З - хлорпропана, снятый при частоте 60 Мгц, приведен на рис. И-3. В этом спектре не выдерживается точно выведенное выше соотношение интенсивностей пиков; причины отклонений, которые являются вполне закономерными, будут рассмотрены позже. Правило сумм интенсивностей соблюдается, и отклонения от него могут быть обусловлены только погрешностью прибора. [15]
Страницы: 1 2