Каприловый спирт
Cтраница 2
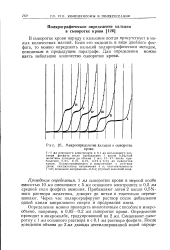 |
Микроопределение кальция в сыворотке крови. [16] |
Прибавляют затем 2 капли 0 5 % - ного раствора желатины, доводят до метки и тщательно перемешивают. Через час полярографируют раствор после добавления одной капли каприлового спирта и продувания азота. [17]
Прибавляют затем 2 капли 0 5 % - ного раствора желатины, доводят до метки водой и тщательно перемешивают. Через час полярографируют раствор после добавления одной капли каприлового спирта и продувания азота. [18]
Радикал октил С8Н17 - широко используется в поверхностно-активных веществах как сам по себе, так и в виде продукта конденсации с ароматическим ядром. Тремя главными источниками этого радикала являются: а) каприловый спирт ( октанол-2), получающийся при пиролизе рицинолеатов, б) 2-этилгексанол, образующийся чисто синтетическим путем из ацетальдегида, и в) диизобутен, получающийся путем димеризации бутена или обработкой изобути-лового или третичного бутилового спирта концентрированной серной кислотой. [19]
Во всех промышленных методах получения себациновой кислоты исходным сырьем служит касторовое масло. При обработке его едким натром приблизительно при 200 С последовательно образуются метил-гексилкетон, ш-оксидекановая кислота и, наконец, себациновая кислота и в качестве главного побочного продукта - каприловый спирт. Температура плавления 88 технической себациновой кислоты 131 С, химически чистой 134 С. [20]
К 1 - 5 мл раствора, помещенного в эрленмейеровскую колбочку на 25 мл, прибавляют 50 - 100 мг цитратного буфера рН 2 5 или 4 7 и 1 каплю каприлового спирта. [21]
Полученную быстро затвердевающую массу нагревают на водяной бане при 40 в течение 5 час. Затем массу растирают в ступке, помещают в прочную круглодонную колбу емкостью 750 - 1000 мл, соединенную с холодильником, и нагревают на вискозиновой бане при 300 - 320 до тех пор, пока не перестанут перегоняться вода и каприловый спирт, на что требуется около 6 - 7 час. Колбу необходимо опускать в масло по горло, которое следует обернуть листовым асбестом. Оставшуюся массу обрабатывают 6 л горячей воды, причем почти вся она растворяется. При охлаждении из раствора выделяются масло и хлопья, которые всплывают и образуют пленку. [22]
В анализируемые растворы, склонные к пенообразова-нию, добавляют 1 каплю или, в случае необходимости, больше каприлового спирта, однако спирт нужно прибавлять в минимальных количествах, так как он может мешать определению реактивом Несслера вследствие образования белого осадка. Нужно следить за тем, чтобы спирт позднее был полностью окислен перекисью водорода при нагревании. Такое же количество каприлового спирта добавляют и в стандартные растворы. Добавляют 0 3 мл серной кислоты 1: 1 и выпаривают воду на электрической плитке. Продолжают нагревание 5 мин после появления белых паров. Снимают колбу и дают остыть 30 сек. Добавляют 0 1 мл 30 % - ной перекиси водорода по стенке колбы, которую держат почти горизонтально. Повторяют обработку перекисью водорода один, четыре или шесть раз, в зависимости от анализируемого объекта. При обычном определении азота в биологических жидкостях и экстрактах обработку перекисью водорода повторяют один раз, при анализе выделенных белков - четыре раза, а при анализе таких стойких веществ, как рибофлавин, тиамин, фолиевая кислота, холин и никотиновая кислота - не менее семи раз. Определения выполняют параллельно на двух пробах при обычной работе и на трех, если необходима большая точность. Охлаждают анализируемый раствор до комнатной температуры. Добавляют 20 мл дистиллированной воды, не содержащей аммиака, смывая ею стенки колбы. Помещают колбу на 5 сек. [23]
Спустя час прибавляют 9 мл насыщенного раствора КгСОз и отгоняют аммиак током воздуха в титрованную кислоту. Для понижения вспенивания прибавляют каприловый спирт. [24]
Для определения мочевины отвешивают 0 5 г пробы и растворяют в 250 сма воды. Прибавляют 10 сма нейтральной уреазы и оставляют на один час, держа горло склянки закрытым. Две или три капли каприлового спирта или петролейного эфира прибавляют к раствору для предотвращения вспенивания. [25]
Добавляют 2 мл концентрированной соляной кислоты и интенсивно кипятят в течение 1 мин. После охлаждения добавляют каплю каприлового спирта и 3 - 4 г бикарбоната натрия в нескольких порциях. По прекращении выделения углекислоты встряхивают смесь в течение 5 мин. Встряхивание не должно быть слишком интенсивным во избежание образования эмульсии. Оставляют до разделения слоев, затем последовательно промывают эфирный слой 25 мл раствора углекислого натрия и 25 мл дистиллированной воды. При образовании стойкой эмульсии после последнего промывания добавляют безводный сернокислый натрий в количестве, достаточном для ее ликвидации. [26]
Осадок сернокислого бария тщательно промывают 3 - 4 раза горячей водой. Каждый раз осадок растирают и сильно встряхивают, плотно закрывая центрифужный стаканчик пробкой. Для избежания вспенивания добавляют несколько капель каприлового спирта. Прозрачный фильтрат и промывные воды упаривают примерно до 75 мл и переносят в центрифужный стакан на 250 мл. [27]
Высшие спирты могут также быть синтезированы при помощи реакции Гербе. Эта давно известная, но редко применяемая реакция заключается в нагревании до высокой температуры низших спиртов с едким натром. Две молекулы спирта при этом конденсируются друг с другом, выделяя воду и образуя спирт с двойным количеством атомов углерода. Этим методом, например, каприловый спирт был превращен в дикаприловый. Сульфат последнего, сходный по своим свойствам с цетил - и миристилсульфатами, стал в небольших количествах выпускаться недавно в США. [28]
Навеску пробы, содержащую около 50 мг мочевины, помещают в колбу, прибавляют 5 мл воды и, если надо, нейтрализуют. После этого всыпают 0 1 г порошка уреазы, закрывают колбу пробкой и помещают на 15 мин. Затем присоединяют колбу к приемнику, содержащему титрованный раствор кислоты, вводят в колбу 20 мл спиртового раствора едкой щелочи ( 4 г едкого натра и 5 мл каприлового спирта ча 1 л метанола) и смесь аэрируют в течение 15 мин. [29]
Когда выделение водорода прекратится, обратный холодильник заменяют нисходящим и продолжают нагревание. Если до окончания перегонки реакционная смесь почему-либо охлаждалась, то при возобновлении нагревания следует принять одну из вышеприведенных мер предосторожности. Спирт, отгоняющийся вместе с водой, время от времени отделяют, а воду возвращают в перегонный сосуд через делительную воронку, вставленную в резиновую пробку рядом с холодильником. Отгонку регулируют так, чтобы дестиллат стекал частыми каплями, но не струей. Отгонка каприлового спирта продолжается около 12 час. К концу отгонки вместе с алкоголем начинают переходить высококипящие продукты, и выделяется значительное количество газа. Эта стадия работы почти всегда связана с затруднениями, так как вследствие продолжительного нагревания припой на швах дна сосуда плавится и через образовавшиеся щели вытекает некоторое количество мыла. Несмотря на это, перегонку следует продолжать, потому что мыло отчасти забивает щели; все же это не позволяет поднять температуру до нужного предела, так как при усилении нагревания увеличивается течь. Верхний слой полученного сырого алкоголя тщательно отделяют от воды и фракционируют. Фракция, кипящая при 100 - 120, состоит, главным образом, из воды, смешанной с небольшим количеством алкоголя. Главная фракция ( 175 - 185) представляет собой метилгексилкарбинол. После вторичной перегонки получают 190 - 200 г ( 23 - 25 % теоретич. [30]
Страницы: 1 2 3