Влияние - электронная структура
Cтраница 1
Влияние электронной структуры на каталитическую активность окислов изучают различными способами. Во-первых, можно последовательно изменять электронную структуру данного окисла и исследовать его активность в отношении данной исследуемой реакции. Подобные изменения, за которыми легче всего проследить путем измерений электропроводности, можно осуществить предварительной обработкой окисла при повышенных температурах в окислительной или восстановительной атмосфере, или предварительной адсорбцией на нем газа донорного или акцепторного типа. [1]
Рассматривая влияние электронной структуры катализатора на активность, Дауден [33] предположил, что карбиды, а также нитриды и карбонитриды должны быть менее активными в синтезе Фишера - Тропша, чем соответствующие металлы, так как электроны атомов в промежутках кристаллической решетки могут заполнять d - оболочки атомов металла ( поскольку считается, что каталитическая активность переходных металлов в реакциях с участием водорода обусловлена наличием незаполненной с. Для железных катализаторов указанная гипотеза, повидимому, неприемлема, поскольку наблюдаемая активность карбидов, нитридов и карбонитридов обычно выше активности восстановленных исходных катализаторов. Однако нельзя утверждать, что экспериментальные факты противоречат этой гипотезе, так как металлическое железо в условиях синтеза склонно к окислению. Подобно этому селективность нитридов, полученных из восстановленного железа, более выражена, чем селективность восстановленных исходных катализаторов или карбонизированных фаз, поскольку восстановленные и карбонизированные катализаторы окисляются в процессе синтеза. [2]
В работах [391, 400] были также сделаны некоторые общие выводы о влиянии электронной структуры комплекса на процесс его кислотной диссоциации. Оказалось, что в изолированном комплексе величиной, определяющей прочность связи протона, является заряд комплекса. Для изозарядных соединений влияние замещения лигандов внутренней сферы на связь О - Н незначительно и может рассматриваться как эффект второго порядка. Для объяснения особенностей кислотной диссоциации аквокомплексов нужно учитывать изменение всех связей в исходном соединении и в продукте реакции. Особенно существенно то, что на рассматриваемый процесс сильно влияют водородные связи между молекулами воды внутренней сферы и окружения. При этом если варьирование состава комплекса вызывает изменение электронной структуры, способствующее кислотной диссоциации, то эти изменения усиливаются при образовании водородной связи с внешнесфер-ными молекулами воды, причем такое усиление происходит и в исходном комплексе, и в продукте диссоциации. Указанные соображения находят известное подтверждение в относительно слабой направленности влияния лигандов на кислотные свойства координированной воды, а также в том факте, что изменения рКа комплексов [ PtL1L2L3OH2 ] n превышают изменения рКс для [ PtLjLaLgCl ] 1 в ряду однотипных объектов. [3]
В работах К - Хауффе [269, 460] в развитие его предыдущих исследований дается анализ влияния электронной структуры примесных полупроводников и образующегося граничного слоя на скорость и механизм реакций. Хауффе получает выражения скорости реакции, содержащие в экспоненте величины, зависящие от изменений уровня Ферми и разности потенциалов граничного слоя и объема полупроводника, и содержащие также величины, являющиеся функциями парциальных давлений реагирующих веществ. На основании выражений скорости процесса и анализа электронных свойств полупроводников в этих работах сделаны предположения о механизме реакций - например разложения закиси азота на р - и - полупроводниках. [4]
К области вопросов, которые следует рассматривать при решении данной задачи, относятся: влияние геометрической и электронной структуры молекул на стехиометрию процесса совместной адсорбции, расчет степени заполнения поверхности молекулами различных типов, ориентация молекул на поверхности адсорбента, проверка гипотез о мономолекулярном ( полимолекулярном) заполнении поверхности адсорбента и ряд других. [5]
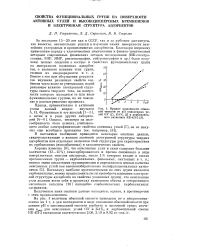 |
Кривые зависимости обменной емкости от рН окисленных углей ОУ ( 7, БАУ0 ( 2 и карбоксильного катионита КБ-4П-2 ( 3. [6] |
Поскольку наиболее протоногенными из этих групп являются карбоксильные, можно предположить ( если пренебречь влиянием электронной структуры адсорбента на свойства указанных групп), что окисленные угли должны вести себя в процессах катионного обмена и гетерогенного кислотного катализа [10] наподобие слабокислотных карбоксильных катионитов. [7]
Многочисленные данные по влиянию строения молекул ингибитора на их защитные свойства можно условно разделить на 2 группы: 1) влияние химической структуры молекул на их защитные свойства, 2) влияние электронной структуры молекул на их защитные свойства. [8]
Кроме учета структуры воды последовательная теория гидратации должна учитывать электронную структуру ионов. Учет влияния электронной структуры ионов рассматривался Ю. А. Кругляком [35 ], Н. А. Измайловым [29, 30] и др. Такие расчеты требуют привлечения квантовой теории. Предполагается, что на основании донорно-акцептор-ного механизма ион образует с первой гидратной оболочкой квазимолекулярную структуру. [9]
Завершение заполнения d - зоны при переходе от Ni к Си приводит к снижению активности. Эта зависимость обусловлена влиянием электронной структуры металла на характер и энергию поверхностного взаимодействия с водородом. [10]

В настоящее время накоплен значительный экспериментальный материал, показывающий влияние природы и положения заместителей в молекуле ингибитора на его защитные свойства. Необходимо отметить, однако, что изложенные представления о влиянии электронной структуры органических соединений на их защитные свойства еще далеко не в полной мере объясняют все аспекты ингибирования и имеют ряд недостатков. [12]
Основная часть исследований выполнена на гранях ( III) монокристаллов иридия и серебра, имеющих ГЦК-структуру с - близкими значениями параметров элементарной ячейки. III) позволяет надежно исключить возможное влияние структурна: факторов к рассмотреть влияние электронной структуры металла на характеристики связи адсорбат-металл. [13]
Изучалась кинетика парофазного гидрирования метилацетилена в статической системе интервале 20 - 230 С в присутствии различных порошкообразных и нанесенных на пемзу катализаторов: никеля, кобальта, железа, родия, иридия, осмия, платины, меди, а также на никель-медных сплавах разного состава. Результаты сходные с полученными ранее для реакции гидрирования ацетилена, обсуждены в аспекте влияния электронной структуры на каталитические свойства металлов и сплавов. [14]
Как видно из приведенных примеров, клешневидные комплексные соединения характеризуются обычно медленным протеканием реакций. Однако у обоих типов соединений образование и распад комплексов с несколькими клешневидными циклами является существенным кинетическим фактором, равноценным влиянию электронной структуры. [15]
Страницы: 1 2