Измерение - основность
Cтраница 1
Измерения основности широко применяются для определения преобладающей структуры потенциально таутомерных соединений. [1]
Вода - стандартный растворитель для измерения основности, но многие вещества в ней недостаточно хорошо растворимы. Недостатком воды является сильная сольватация ею нейтральной и протонированной азагруппы. Поэтому более предпочтительны для работы апротонные растворители, например ацетонитрил. К сожалению, до сих пор проведено относительно мало измерений величины рКа в ацетонитриле. Данные табл. 5.5 показывают, что в ацетонитриле основность гетероциклов по сравнению с водой увеличивается на 7 - 8 порядков. [2]
Бейкер подкрепляет свою аргументацию результатами измерений основности, но основности тиоловых сложных эфиров очень близки к основностям соответствующих кислородсодержащих сложных эфиров, несмотря на то что карбонильные частоты являются более низкими. Такой подход не дает однозначного решения при выборе между двумя альтернативными резонансными формами. Беллами [38] указывал, что основность определяется доступностью неподеленной пары электронов атома кислорода и что эти электроны не обязательно затрагиваются при эффектах делокализации, в которых принимают участие л-электроны кратной связи. Так как в последнем случае увеличение происходит вследствие эффекта конкуренции между циклом и карбонильной группой за неподеленную пару электронов атома кислорода, то представляется вероятным, что аналогичная схема резонанса имеет место и в случае тиоловых сложных эфиров. [3]
В зависимости от реакционной серии и диапазона измерения основности Y - может преобладать то или иное влияние и наблюдаться ускорение ( 50) или замедление ( р0) реакций с ростом основности Y -, что показано на рис. 34 для реакции этиленоксида с фенолами при катализе соответствующими фенолятами. [4]
В табл. 21а приведена большая часть литературных данных по различным способам измерения основности простых эфиров. Для наглядного сравнения этих методов в табл. 5 приведены качественные ряды основности нескольких особенно важных соединений по отношению к некоторым кислотным системам. [5]
Значительное число тиоловых сложных эфиров было исследовано Найквистом и Поттсом [67]; Бейкер и Харрис [48] изучили меньше этих соединений, однако они выполнили измерения основности. [6]
Основность гетероциклического атома азота зависит от плотности электронов у этого атома; так, изменения электронной плотности, обусловленные присутствием различных заместителей, можно определить путем измерения основности соответствующих соединений. Значения рКа для пиридинов, азинов и азолов уже рассматривались ( стр. [7]
Аминопиридины ( пример: 609) и соответствующие амины - бензопиридинов могут также находиться в таутомерном равновесии с пиридониминной формой ( 611, 612), но измерение основности ( ср. Это равновесие можно сравнить с таутомерией 2 - и 4-оксипиридинов, которые существуют главным образом в пиридонной форме ( см. стр. [8]
Они изучили около 32 моно-и дизамещенных азобензолов в качестве индикаторов в 20 % - ном водно-этанольном растворе серной кислоты с целью создания функции кислотности, которая была бы связана с водной шкалой Я0 посредством измерений основностей п - и о-нитроанилинов в тех же условиях. Превосходное совпадение значений Я0 для различных членов рядов говорит о том, что для молекул близкого строения в этой среде может существовать хорошая функция кислотности. [9]
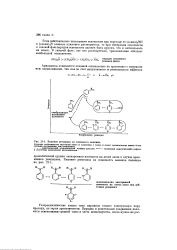 |
Влияние резонанса на основность анилина. [10] |
Если действительно уменьшение основности при переходе от ( алкил) а. ЧН к ( алкил) 31Ч вызвано влиянием растворителя, то при измерении основности в газовой фазе порядок основности должен быть другим, что и наблюдалось на опыте. В газовой фазе, где нет растворителя, триалкиламин обладает наибольшей основностью. [11]
Можно предположить, что с изменением состава водных растворов серной кислоты относительное количество различных агрегатов [115, 116] свободной воды ( основания, которое мы изучаем) также изменяется. Образно говоря, мы стреляем в непрерывно движущуюся и изменяющуюся мишень, поскольку соединения, которые хотелось бы представить как свободное основание и его сопряженную кислоту, очень многочисленны и претерпевают непрерывные изменения по мере изменения самой кислотности, необходимой для измерения основности. [12]
Смещения при этом достигают 70 - 150 см-1 в зависимости от выбранных карбонильного соединения и льюисовой кислоты. Лепперт [111] использовал значения vCO таких соединений для получения количественной оценки акцепторных свойств кислот. Возможность обратного подхода - использования стандартной кислоты и измерения основности карбонильных соединений - не была систематически исследована, но есть много отрывочных данных, которые демонстрируют применение этого метода. [13]
Более удивительно на первый взгляд, что описанное простое соотношение частота - основность не выполняется для сопряженных карбонильных соединений. Понижение vCO при сопряжении почти наверняка обусловлено реальным изменением силовой постоянной карбонильной связи, но это изменение никак не отражается на основности, которая, например, остается фактически неизменной при переходе по ряду ацетон-ацетофенон-бензофенон. Высказывалось предположение, что это происходит потому, что результаты измерений основности определяются состоянием гибридизации неподеленной пары электронов кислородного атома и что оно не обязательно будет меняться при делокализации л-электронов в плоскости, перпендикулярной связи. Это также указывает на то, что просто карбонильная частота или силовая постоянная сами по себе не могут однозначно характеризовать распределение электронной плотности по связи. Два соединения вполне могут иметь одни и те же силовые постоянные и частоты колебаний карбонильной связи, но в одном случае vCO может быть повышена вследствие небольшого индукционного эффекта, а в другом изменена за счет значительно большего индукционного эффекта, который частично компенсируется резонансным эффектом. Частоты при этом будут одни и те же, а распределение электронной плотности различное. Следует еще раз отметить, что надежные количественные предсказания могут быть сделаны только для близких родственных соединений. Попытки сопоставления карбонильных частот соединений различных типов, вероятнее всего, приведут к ошибочным результатам, если только не принимать во внимание второй параметр - основность. Если для пары соединений совпадают частоты и основности, то можно с уверенностью утверждать, что у них подобно и распределение электронной плотности, но любой из этих параметров может изменяться при неизменном другом параметре. По-видимому, наиболее целесообразно сочетать измерения обоих типов для окончательной количественной оценки роли различных химических факторов, влияющих на групповые частоты. [14]
Трудно точно знать, какое свойство измеряется при этом, поскольку взаимодействие кислоты с реакционноспособными соединениями в бензоле является взаимодействием, сопровождающимся образованием водородной связи, или в лучшем случае дает агрегаты ионов. Влияние основания на эту реакцию должно иметь довольно сложный характер. Однако в целом такие данные плохо коррелируются с другими данными по измерению основности. Кроме того, если для основания построить график зависимости логарифмов факторов замедления двух реакций, катализируемых кислотой, друг от друга, то получится беспорядочный набор точек. Это означает, что метод не имеет самостоятельного значения. Мы включили в приведенные в конце статьи таблицы большинство этих данных, предполагая, что ими можно пользоваться только для сравнения близких родственных соединений, и то с осторожностью. [15]
Страницы: 1 2