Измерение - площадь - пятно
Cтраница 3
Ввиду того, что пролин хорошо отделяется от других аминокислот на бумаге при помощи хроматографии / 1 / или электрофореза, желательно использовать эту возможность при количественном его определении. Предлагаемый иэатиновый метод / Т / количественного определения пролина измерением площади пятен недостаточно точен. Кроме того, при проявлении хроматограммы изатином не определяются остальные аминокислоты, проявляемые нингид иновым реактивом. [31]
Метод ТСХ применяется для определения в воде следовых количеств металлов. Количественное определение кобальта, свинца и магния основано на измерении площадей пятен на хромато-грамме. [32]
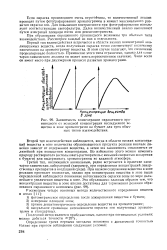 |
Зависимость концентрации окрашенного производного от исходной концентрации исследуемого вещества в зоне хроматограмм на бумаге для трех обычных типов взаимодействия. [33] |
Если окраска производного очень неустойчива, то количественный анализ проводят путем фотографирования хроматограммы в момент максимального развития окраски зон. Для повышения контрастности при фотографировании используют соответствующие светофильтры. Далее количественный анализ выполняют путем измерения площади пятен или денситометрически. [34]
Следует подчеркнуть, что изложение в данном разделе ограничено положениями, справедливость которых связана с неоднократно упоминаемыми предположениями. На практике в каждом случае необходима критическая проверка. Упоминавшаяся статья Гиддингса [80] содержит литературный обзор опытных данных по измерению площади пятен на бумажных хромато-граммах. Результаты из области хроматографии в тонких слоях описаны в гл. [35]
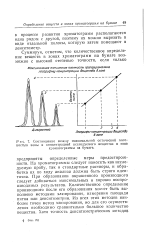 |
Соотношение между максимальной оптической плотностью зоны и концентрацией исследуемого вещества в зоне хроматограммы на бумаге. [36] |
На хроматограммы следует наносить как исследуемую пробу, так и стандартные растворы, и обработка их по ходу анализа должна быть строго идентична. При образовании окрашенного производного следует добиваться максимальных и хорошо воспроизводимых результатов. Количественное определение производного после его образования может быть выполнено методами элюирования, измерения площади пятна или денситометрии. Из всех этих методов наиболее точен метод элюирования, но для его осуществления необходимо сравнительно большое количество вещества. [37]
Особенно подробно изложены в книге вопросы1 теории и техники двух основных методов количественного определения веществ в зонах: метода, предусматривающего элюирование, и методов прямого ( без - отделения сорбента-носителя) определения веществ в зонах хроматограмм. Последние методы более чувствительны, менее трудоемки и поэтому весьма перспективны при анализе микро - и субмикроколичеств смесей. Рассмотрены три главных способа количественного определения веществ непосредственно на хро-матограммах: визуальное сравнение интенсивности окраски зон, измерение площади пятна и денситометрия. [38]
Предложен метод раздельного определения 2 4 - Д кислоты и бутилового эфира в воде с помощью хроматографии в тонком слое силикагеля. В качестве подвижной фазы применяют циклогексан бензол ледяная уксусная кислота. Хромато-граммы обрабатывают реактивом, содержащим AgNO, и проявляют в УФ-свете. Количественное определение проводят по измерению площади пятен. [39]
Полученные хроматограммы трудно хранить из-за малой механической прочности слоя, поэтому после проявления их при необходимости фотографируют и хранят фотокопии. Оригинальную хроматограмму можно сохранить, если законсервировать слой 4 % - ным раствором коллодия, к которому добавлено 7 5 % глицерина. Иногда для фиксирования результатов анализа снимают с хроматограммы точную копию на кальку, просветленную вазелиновым маслом. Эти копии целесообразно использовать для измерения площадей пятен веществ-свидетелей и пробы. [40]
Количественный анализ с помощью метода ТСХ проводился [95-97] при определении содержания ВМА, ВНТ, пропилгал-лата ( PG) и нордигидроваяретовой кислоты ( NDGK. По окончании разделения он элюировал соединения с пластинки с выходами 82 - 101 % и определял содержание антиоксидантов колориметрически. Амано и др. [96] определяли содержание ВНА в пищевых маслах методом измерения площади пятен. Масла перегоняли с водяным паром и экстрагировали антиоксидант из дистиллата тетрахлоридом углерода. [41]
Страницы: 1 2 3